
Węglowce - są to pierwiastki 14 (daw. IVA lub IV głównej) grupy układu okresowego. Są to węgiel (C), krzem (Si), german (Ge), cyna (Sn), ołów (Pb) i flerow (Fl).
Odkrycie węglowców: Trzy z sześciu węglowców znano już w starożytności. Były to węgiel, cyna i ołów. Krzem i german zostały odkryte w XIX wieku (krzem przez Antoine Lavoisiera, zaś german przez Clemensa Alexandera Winklera). Flerow zostł otrzymany sztucznie w 1999 roku przez naukowców z Instytutu Badań Jądrowych w Dubnej.


(...)
Występowanie węglowców w przyrodzie: Węgiel wchodzi w skład licznych związków organicznych. Ponadto występuje w przyrodzie w postaci węgli kopalnych. Najważniejszym minerałem zawierającym węgiel jest węglan wapnia. Dużo rzadziej spotyka się go w postaci diamentów. W środowisku naturalnym wyjątkowo daje się odnaleźć w formie fulerenów (np. w strzałkach piorunowych).

Krzem zajmuje drugie miejsce pod względem zawartości w przyrodzie (zaraz po tlenie). Nie występuje jednak w stanie wolnym, ale w postaci krzemionki, krzemianów i glinokrzemianów.
German występuje w przyrodzie w niewielkich ilościach, tylko w postaci związków, np. minerałów takich jak germanit (Cu2GeS3) i argyrodyt (Ag8GeS6). Niewielkie ilości germanu są zawarte w węglu kamiennym.
Głównym źródłem cyny są minerały: kasyteryt (SnO2), stannin (Cu2FeSnS4).





(...) Ołów występuje w niewielkich ilościach w postaci rodzimej, jednak jego głównym źródłem jest galena (PbS).



(...) Flerow jest syntetycznym pierwiastkiem promieniotwórczym i nie występuje na Ziemi.
Właściwości węglowców: Ze wzrostem liczby atomowej w tej grupie maleje wpływ biernej pary elektronowej. Wynikiem tego jest malejąca trwałość pierwiastków na IV stopniu utlenienia i rosnąca na II. Wszystkie z nich oprócz ołowiu tworzą struktury diamentu, przy czym w przypadku węgla odmiana ta jest izolatorem, a dalsze pierwiastki są, z powodu malejącej szerokości pasma wzbronionego, półprzewodnikami i przewodnikami.

(...) Węgiel jest ciałem stałym, nierozpuszczalnym w wodzie. Grafit jest czarnoszary, odpowiednio wypolerowany ma metaliczny połysk. Diament jest bezbarwny, bezwonny, nie ma smaku. Jest pierwiastkiem mało reaktywnym; w wysokiej temperaturze reaguje z fluorem, tlenem, siarką i metalami 1, 2 i 13 grupy. Rozżarzony koks reaguje z wodą, dając mieszaninę tlenku węgla i wodoru (tzw. gaz syntezowy): C + H2O → CO + H2.


Tworzy trzy odmiany alotropowe:
- Diament – bezbarwny, krystaliczny, ma dużą gęstość i twardość; podczas ogrzewania do temperatury powyżej 1500 °C przechodzi w grafit.
- Grafit – mała twardość, łupliwość; jest bardziej reaktywny niż diament.
- Fulereny – czarne ciała stałe o metalicznym połysku, odpowiednio domieszkowane mają własności nadprzewodzące i półprzewodnikowe.
W związkach chemicznych występuje na -IV, II i IV stopniu utlenienia. Krzem to szare, błyszczące, kruche ciało stałe. Jest półprzewodnikiem. W związkach jest czterowartościowy. Występuje, podobnie jak węgiel, w strukturze diamentu. German jest kruchym, srebrzystobiałym półmetalem o właściwościach półprzewodnikowych. Nie reaguje z wodą i kwasami (oprócz kwasu azotowego).
Cyna jest ciałem stałym. Występuje w trzech odmianach krystalicznych:
- Cyna α – ma strukturę diamentu, która ma właściwości półprzewodnikowe; występuje jako proszek barwy szarej; przemiana w cynę β zachodzi powoli w temperaturze 13,2 °C i wyższej;
- Cyna β – srebrzystobiała z niebieskawym odcieniem;
- Cyna γ.

(...) W związkach chemicznych cyna występuje na -IV, II i IV stopniu utlenienia. Ołów to szarosrebrzysty,miękki, kowalny metal. Na powietrzu pokrywa się warstwą tlenku. Jego sole są zazwyczaj słabo rozpuszczalne lub nierozpuszczalne w wodzie (z wyjątkiem azotanu ołowiu (II) i octanu ołowiu (II)).
Właściwości węglowców w związkach z wodorem:
Związki wodoru z:
- węglem to węglowodory;
- krzemem silany;
- germanem germanany;
- cyną stannany;
- ołowiem plumbany.
W szeregu od węgla do ołowiu maleje trwałość tych związków. Węgiel tworzy najwięcej związków z wodorem (węglowodory) dzięki trwałym wiązaniom węgiel-węgiel (katenacja). Są one trwałe i jako jedyne z tej grupy nie hydrolizują w wodzie. Pierwszy krzemowodór (SiH4) jest nietrwały w środowisku wodnym i na powietrzu.


Najważniejsze związki chemiczne węglowców
Węgiel:
- Dwutlenek węgla (CO2)
- Tlenek węgla (CO)
- Kwas węglowy (H2CO3)
- Węglik wapnia (CaC2)
- Cyjanowodór (HCN)
- Związki organiczne
Krzem:
- Dwutlenek krzemu (SiO2)
- Kwas krzemowy (H2SiO3)
- Kwas ortokrzemowy (H4SiO4)
- Krzemiany
- Krzemowodory
- Krzemki
- Węglik krzemu (karborund) (SiC)
German:
- Germanowodory
- Siarczek germanu(II) (GeS)
- Siarczek germanu(IV) (GeS4)
- Tlenek germanu(II) (GeO)
- Tlenek germanu(IV) (GeO2)
Cyna:
- Tlenek cyny(II) (SnO)
- Tlenek cyny(IV) (SnO2)
- Kwas cynowy ((SnO2)x•(H2O)y)
- Wodorotlenek cyny(II) (Sn(OH)2)
- Cynowodór (SnH4)
- Siarczek cyny(II) (SnS)
- Siarczek cyny(IV) (SnS2)
- Chlorek cyny(II) (SnCl2)
- Chlorek cyny(IV) (SnCl4)
Ołów:
- Węglan ołowiu(II) (PbCO3)
- Chlorek ołowiu(II) (PbCl2)
- Chlorek ołowiu(IV) (PbCl4)
- Chromian ołowiu(II) (PbCrO4)
- Azydek ołowiu(II) (Pb(N3)2)
- Azotan ołowiu(II) (Pb(NO3)2)
- Tlenek ołowiu(II) (PbO)
- Tlenek ołowiu(IV) (PbO2)
- Tlenek diołowiu(II), ołowiu(IV) (Pb3O4)
- Wodorotlenek ołowiu(II) (Pb(OH)2)
- Siarczek ołowiu(II) (PbS)
- Siarczan ołowiu(II) (PbSO4)
Otrzymywanie: Węgiel do celów laboratoryjnych otrzymuje się przez prażenie sacharozy bez dostępu tlenu z powietrza. Na skalę przemysłową otrzymuje się go z węgli kopalnych, przez rozkład termiczny drewna oraz jako diament.
Krzem można otrzymać w laboratorium przez redukcję krzemionki magnezem: SiO2 + 2 Mg → 2 MgO + Si. Na skalę przemysłową redukuje się krzemionkę węglem (SiO2 + C → CO2 + Si) lub węglikiem wapnia.
Pozostałe trwałe węglowce są otrzymywane przez redukcję ich tlenków. Ołów można otrzymać elektrolitycznie z jego siarczku. Flerow można uzyskać wyłącznie w reakcjach jądrowych.
Zastosowanie węglowców: Węgiel jest podstawowym składnikiem związków organicznych. Diament służy do wyrobu biżuterii. Ze względu na jego twardość używa się go także do wyrobu narzędzi do obróbki stali i szkła. Grafit jest używany do wyrobu elektrod, grafitów do ołówków i tygli laboratoryjnych. Węgle kopalne są wykorzystywane jako surowce energetyczne. Izotop węgla 14C jest stosowany jako wskaźnik izotopowy, zaś 1/12 masy izotopu 12C stanowi wzorzec jednostki masy atomowej. Krzem o dużym stopniu czystości jest stosowany do wyrobu półprzewodników. Jest też stosowany do odtleniania specjalnych gatunków stali i jako składnik wielu stopów. Krzem i jego związki są surowcami w przemyśle szklarskim, ceramicznymi materiałów budowlanych. German stosuje się do produkcji półprzewodników, luminoforów, filtrów optycznych i stopów specjalnych.


(...) Cyna jest używana do pokrywania metali mniej odpornych na korozję. Jest też składnikiem stopów. W średniowieczu wykonywano wiele przedmiotów z cyny, ze względu na jej dostępność i niską cenę.
Ołów służy do wyrobu rur kanalizacyjnych i ekranów chroniących przed promieniowaniem. Wykłada się nim także komory do produkcji kwasu siarkowego, celulozy i wapna bielącego. Ołowiu używa się też do wyrobu płyt akumulatorowych, szkła ołowiowego i do otrzymywania innych związków ołowiu.







 1) Węgiel (C, łac. carboneum) - jest to pierwiastek chemiczny o liczbie atomowej 6, niemetal z bloku p układu okresowego. Należy do grupy 14. Ma cztery elektrony walencyjne. Istnieją trzy naturalnie występujące izotopy węgla, 12C oraz 13C są stabilne, natomiast izotop 14C jest promieniotwórczy o czasie połowicznego rozpadu równym około 5700 lat[8]. Węgiel jest jednym z niewielu pierwiastków znanych w starożytności. Jako pierwszy polską nazwę – węgiel – zaproponował Filip Neriusz Walter.
1) Węgiel (C, łac. carboneum) - jest to pierwiastek chemiczny o liczbie atomowej 6, niemetal z bloku p układu okresowego. Należy do grupy 14. Ma cztery elektrony walencyjne. Istnieją trzy naturalnie występujące izotopy węgla, 12C oraz 13C są stabilne, natomiast izotop 14C jest promieniotwórczy o czasie połowicznego rozpadu równym około 5700 lat[8]. Węgiel jest jednym z niewielu pierwiastków znanych w starożytności. Jako pierwszy polską nazwę – węgiel – zaproponował Filip Neriusz Walter.
Znanych jest kilka odmian alotropowych węgla, z czego najbardziej znane to grafit oraz diament. Właściwości fizyczne węgla zależą od odmiany w jakiej występuje. Na przykład diament jest przezroczysty, natomiast grafit jest nieprzezroczysty i czarny.
Pol: Wykaż różnice we właściwościach fizycznych grafitu i diamentu.

(...) Diament jest jednym z najtwardszych materiałów na Ziemi, podczas gdy grafitem można narysować kreskę na papierze. Diament ma bardzo niskie przewodnictwo właściwe, a grafit jest dobrym przewodnikiem elektrycznym.





(...) Diament ma najwyższą przewodność cieplną ze wszystkich znanych materiałów w warunkach normalnych. Wszystkie odmiany alotropowe węgla są w warunkach normalnych ciałami stałymi. Innymi odmianami alotropowymi węgla są: fuleren oraz formy poliynowe. Niektórzy uważają też, że jego odmianami alotropowymi są: nanocebulka, nanorurka, nanopianka, karbin, choć są to raczej nazwy struktur supramolekularnych, niż odmiany alotropowe w pełnym tego słowa znaczeniu.




(...) Wszystkie formy występowania węgla są wysoce stabilne, wymagają wysokiej temperatury żeby przereagować nawet z tlenem. Największe ilości nieorganicznego węgla występują w postaci skał wapiennych, dolomitów oraz dwutlenku węgla, natomiast znaczne ilości węgla organicznego znajdują się w paliwach kopalnych. Węgiel tworzy więcej związków niż wszystkie inne pierwiastki chemiczne. Liczba organicznych związków węgla zarejestrowanych w bazie Beilstein w roku 2008 wynosiła 10 853 341, jednak liczba jego potencjalnych związków jest nieograniczona.
Węgiel znajduje się na czwartym miejscu najczęściej występujących pierwiastków we Wszechświecie, po wodorze, helu i tlenie. Jest obecny we wszystkich organizmach żywych. W ludzkim ciele jest po tlenie najliczniejszym pierwiastkiem ze względu na masę (ok. 18,5%). Ta ilość w połączeniu z różnorodnością związków organicznych stawia węgiel jako chemiczną podstawę życia.



Źródło: atomfortheworld.blogspot.com





Charakterystyka węgla: Różne odmiany alotropowe węgla wykazują bardzo różne właściwości, np. diament jest najtwardszą naturalnie występującą substancją, grafit jest jedną z substancji o najmniejszej twardości. Ponadto węgiel ma powinowactwo do tworzenia wiązań chemicznych z innymi małymi atomami, włączając w to inne atomy węgla oraz tworzenia wielu wiązań kowalencyjnych z tymi atomami w wyniku czego związki zawierające węgiel w swojej strukturze stanowią znaczną cześć wszystkich znanych związków, liczba ich dochodzi do dziesięciu milionów[13]. Węgiel ma także najwyższą temperaturę topnieniaze wszystkich pierwiastków[14]. Przy ciśnieniu atmosferycznym nie występuje w stanie ciekłym, lecz podczas ogrzewania sublimuje w temperaturze 3852 °C; jego punkt potrójny występuje przy ok. 10,3 MPa (102 atm)[1]. Niezależnie od odmian alotropowych pozostaje ciałem stałym w wyższych temperaturach, niż metale o najwyższych temperaturach topnienia (wolfram i ren). Jednakże termodynamicznie węgiel jest podatny na utlenianie, znacznie bardziej niż żelazo czy miedź, które są słabymi reduktorami w temperaturze pokojowej.
Związki zawierające węgiel są podstawą życia na Ziemi, a cykl węglowo-azotowo-tlenowy dostarcza część energii wytwarzanej przez Słońce i inne gwiazdy.




(...) Pomimo różnorodności związków węgla większość jego form jest stosunkowo słabo reaktywna w warunkach normalnych. Nie reaguje z kwasem siarkowym, kwasem solnym, chlorem ani zasadami. W podwyższonej temperaturze węgiel reaguje z tlenem, tworząc tlenki węgla, oraz redukuje wiele tlenków metali, takich jak tlenek żelaza, do wolnego metalu. Ta reakcja egzotermiczna jest wykorzystywana w przemyśle żelaza i stali do kontroli zawartości węgla w stali:
- Fe3O4+ 4C(s) → 3Fe(s) + 4CO(g)↑
Z siarką tworzy dwusiarczek węgla, a z parą wodną tlenek węgla i wodór:
- C(s) + H2O(g) → CO(g) + H2(g)
Węgiel reaguje z niektórymi metalami, tworząc węgliki, takie jak węglik żelaza (cementyt) czy węglik wolframu, który dzięki swojej twardości jest używany w różnego rodzaju narzędziach tnących.


Katenacja (od łac. catenatus, im. czas. catenare, od catena, „łańcuch”) - tworzenie wiązań między atomami tego samego pierwiastka, prowadzące do powstania łańcuchowych związków chemicznych. Katenacja występuje tylko w przypadku pierwiastków mających wartościowość co najmniej 2, mogących tworzyć między sobą silne wiązania chemiczne. Zdolność do katenacji charakteryzuje przede wszystkim atomy węgla, natomiast mniejsze znaczenie ma w przypadku siarki i krzemu. Rzadko występuje dla germanu, azotu, selenu i telluru.

narzędziach do skrawania, zwłaszcza szlifowania i wiercenia. Węgiel bezpostaciowy jest używany w medycynie oraz jako węgiel aktywny do procesów filtracji i oczyszczania.

Zagadnienia związane z węglem:
*Aerografit – najlżejszy materiał na świecie opracowany przez uczonych z Uniwersytetu Technicznego Hamburg-Harburg oraz Uniwersytetu Christiana-Albrechta w Kilonii.
Gęstość aerografitu wynosi 0,2 mg/cm3. Zbudowany jest z siatki pustych w środku węglowych rurek, dzięki czemu może przenieść obciążenie 40 000 razy większe od swojej masy. Jest dobrym przewodnikiem prądu elektrycznego.






Willard Frank Libby (ur. 17 grudnia 1908, zm. 8 września 1980) – chemik amerykański, laureat Nagrody Nobla w dziedzinie chemii za rok 1960.
 |
| Źródło: thefamouspeople.com - Willard Frank Libby. |
Młodość i edukacja: Urodził się 17 grudnia 1908 w Grand Valley, w stanie Kolorado. Jego ojciec, Ora Edward Libby, był rolnikiem, edukację zakończył na trzeciej klasie szkoły podstawowej. Matką Willarda Franka była Eva May Rivers. W 1913 rodzice przenieśli się do północnej Kalifornii i zajęli się sadownictwem. Willard ukończył szkołę średnią w 1926. Zachęcany przez rodziców, kształcił się dalej na Uniwersytecie Kalifornijskim w Berkeley. Początkowo zamierzał zostać inżynierem górnikiem, później zainteresowała go chemia, matematyka i fizyka. Studia ukończył w 1931.
W ciągu dwóch lat po uzyskaniu doktoratu Libby prowadził badania w dziedzinie niskich energii i promieniotwórczości. W tym czasie zbudował czuły licznik Geigera do wykrywania słabego promieniowania. W latach 1933-1940 wykładał na Uniwersytecie w Berkeley.
Projekt Manhattan: W czasie II wojny światowej Libby przeniósł się do War Research Division przy Uniwersytecie Columbia i w ramach Projektu Manhattan prowadził badania nad energią atomową. Wziął udział w opracowaniu sposobu rozdzielania izotopów uranu, potrzebnych do uzyskania bomby atomowej. To ważne osiągnięcie opierało się na zasadach, które Libby później wykorzystał w pracach nad datowaniem za pomocą węgla promieniotwórczego C-14.
Wynalezienie datowania radiowęglowego: Libby wynalazł metodę mierzenia czasu połowicznego rozpadu izotopu węgla 14C, co stało się podstawą bardzo ścisłego datowania próbek, tzw. datowania radiowęglowego, zwłaszcza pochodzenia organicznego (zwłok, szkieletów, papirusów, skamielin itd.). Metoda ta znalazła zastosowanie we współczesnej archeologii i kryminologii. W 1960 r. Libby otrzymał za jej opracowanie Nagrodę Nobla w dziedzinie chemii.
Już wcześniej zauważono, że rozpad promieniotwórczy dostarcza informacji na temat wieku Ziemi. Po kilku latach od odkrycia promieniotwórczości stwierdzono, że rozpad jądra prowadzi do przekształcenia nietrwałych pierwiastków promieniotwórczych w trwałe, zwykłe pierwiastki, przy czym czas rozpadu jest charakterystyczny dla danego izotopu i można go oznaczyć. Już w 1904 r. Ernest Rutherford doszedł do wniosku, że zjawisko to może posłużyć do określenia wieku Ziemi. W rok później tym problemem zajął się amerykański chemik Bertram Borden Boitwood. W 1907 r. opracował metodę, dzięki której ustalił wiek Ziemi na 2,2 mld lat, a wiek Układu Słonecznego co najmniej na 5 mld lat.
Ważne dla sprawdzenia tej hipotezy okazało się uwzględnienie możliwości, jakie zawierały wykryte w 1939 r. promieniowanie kosmiczne bombardujące Ziemię. Promieniowanie kosmiczne to strumień cząstek elementarnych stale docierających z przestrzeni kosmicznej na Ziemię. Cząstki zawarte w promieniowaniu kosmicznym po drodze zderzają się i wchodzą w reakcję z cząsteczkami azotu. W 1946 Libby wykazał, że pewna część atomów azotu zmienia się w promieniotwórczy węgiel, czyli w izotop węgla C-14, a ten z kolei szybko przechodzi w dwutlenek węgla, przyswajany przez rośliny i obecny w całej przyrodzie. Wobec tego wszystkie organizmy żywe pochłaniają naturalny C-14 wraz z pożywieniem.
Libby wysunął hipotezę, że zasadne jest przypuszczenie, iż zawartość C-14 w organizmach żywych pozostaje stosunkowo niezmienna w ciągu całego ich życia, to znaczy tak długo, jak długo przyswajają one składniki odżywcze. Po obumarciu organizmu izotop C-14, znajdujący się w roślinach i zwierzętach, rozpada się, a jego zawartość z czasem się zmniejsza. Czas połowicznego rozpadu uranu U-238 wynosi 4,5 mld lat, a węgla C-14 jak stwierdzono około 1940 r. jest stosunkowo krótki i wynosi zaledwie około 5730 lat.
Libby zbudował specjalny licznik Geigera, zamknięty w grubej ołowianej obudowie, która pochłaniała naturalne promieniowanie z zewnątrz, i opracował metodykę pomiarów. Najpierw sprawdził metodę na roślinach, między innymi na sekwojach kalifornijskich, których wiek ustalono w inny sposób. Później oznaczył wiek przedmiotów wykonanych z drewna – na przykład łodzi pogrzebowej faraona Egiptu Senusreta – lub pozostałości organicznych i osiągnął doskonałą zgodność między wiekiem przewidywanym a ustalonym doświadczalnie.
Zdołał także ustalić wiek najstarszych wytworów człowieka oraz ludzkich wspólnot. Na tej podstawie wysunął wniosek, że epoka lodowcowa skończyła się około 10 000 lat temu, dużo później niż przedtem przypuszczano. Datowanie za pomocą węgla C-14 daje dobre wyniki dla okresu od 500 do 70 000 lat temu.
Po II wojnie światowej Libby zajmował znaczącą pozycję w fizyce amerykańskiej. W 1952 r. Libby opublikował książkę Radiocarbon Dating. W latach 1945-1954 był profesorem chemii w Institute for Nuclear Studies na Uniwersytecie Chicagowskim.
Dalsza kariera naukowa: Później wziął urlop, by uczestniczyć w pracach Atomic Energy Commission. Libby, mianowany do tego ciała przez prezydenta Dwighta Eisenhowera, był uważany za zwolennika zimnej wojny i obrońcę polityki rządu.
W latach 50. XX w. gorąco popierał budowę przydomowych „schronów atomowych”, które rzekomo miały uchronić ludzi przed śmiertelnymi skutkami promieniowania powstającego na skutek ataku atomowego. Wykazywał zdumiewający spokój w ocenie zagrożenia promieniowaniem i stanowczo opowiadał się za próbami z bronią jądrową.
Od 1959 ostatnie lata pracy zawodowej Libby spędził na wydziale chemii Uniwersytetu Kalifornijskiego w Los Angeles, był także dyrektorem Institute of Geophysics and Planetary Physics.
Przeszedł na emeryturę w 1976. Zmarł 8 września 1980 w wyniku komplikacji wywołanych zapaleniem płuc[potrzebny przypis].
Znaczenie: Datowanie za pomocą węgla C-14, od czasu odkrycia, przekształciło się w rozległą dziedzinę badań, w których wykorzystuje się też inne izotopy promieniotwórcze, a stosowane metody są coraz bardziej wyszukane i dokładne. Nowe sposoby, na przykład metoda K-Ar z zastosowaniem promieniotwórczego potasu K-40, służą do ustalania wieku kontynentów i struktury geologicznej. Metody Rb-Sr z zastosowaniem atomów rubidu Rb-87 użyto do ustalenia wieku skał pobranych na Księżycu.
Życie osobiste i osobowość: Ożenił się z Leonorą Lucindą Hickey, z którą miał córki bliźniaczki, Susan i Janet. Po rozwodzie w 1966 ożenił się z Leoną Woods Marshall.
Był człowiekiem wysokim i mocno zbudowanym, miał rude włosy. Przez całe życie był znany jako „dziki Bili”. Uważano go za dobrego nauczyciela, lecz bardzo surowego dla doktorantów. Jego poglądy na temat koniecznych cech fizyka były typowe dla okresu, w którym żył.

*Kardyf (od ang. Cardiff – miasto w Walii) – gatunek angielskiego węgla kamiennego, który odznacza się wysokim przewodnictwem cieplnym i łatwopalnością, używany głównie na statkach.







Sapropelit (węgiel sapropelowy) – węgiel kopalny, powstały wskutek diagenezy i metamorfizmu sapropelu. W zależności od składu macerałów wyróżnia się wśród nich boghedy i kennele.

Zawiesina wodno-węglowa – paliwo ciekłe składające się z drobnych ziaren węgla kopalnego zawieszonych w wodzie (często 55-70% węgla, 30-45% wody). Do jej produkcji można stosować zarówno węgiel kamienny, jak i brunatny. Cząstki węgla mają rozmiary rzędu mikrometrów. Stosowana w energetyce (spalanie w kotłach) oraz jako zastępcze paliwo ciekłe (na przykład w silnikach Diesla).
*Nieorganiczne związki węgla:

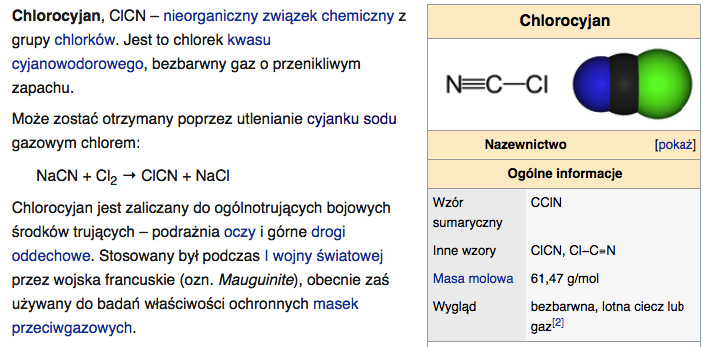


































--> Cyjanki:



















--> Węgliki:








































Izotiocyjaniany (ITC) – sole lub estry kwasu izotiocyjanowego będące tautomerami rodanków (tiocyjanianów), a także analogami siarkowymi izocyjanianów. Zawierają jednowartościowy anion [N=C=S]−
i należą do grupy pseudohalogenków. Niektóre z nich są wykorzystywane przy sekwencjonowaniu białek metodą degradacji Edmana.
Część estrów izotiocyjanianowych powstaje naturalnie podczas hydrolizy glukozynolanów występujących w niektórych roślinach, głównie w warzywach krzyżowych, np. kiełkach brukselki, kapuście, brokułach, jarmużu, wasabi i rzeżusze. Niektóre z tych związków wykazują działanie antykancerogenne.



 2) Krzem (Si, łac. silicium) - pierwiastek chemiczny, z grupy półmetali w układzie okresowym. Izotopy stabilne krzemu to 28Si, 29Si i 30Si. Wartościowość w większości związków wynosi 4, rzadziej spotykane są związki z krzemem dwuwartościowym. Typowe stopnie utlenienia to –IV i IV, rzadko –II i II; znane są też związki o st. utl. 0. Krzem (w postaci monokryształów) jest wykorzystywany powszechnie w przemyśle elektronicznym.
2) Krzem (Si, łac. silicium) - pierwiastek chemiczny, z grupy półmetali w układzie okresowym. Izotopy stabilne krzemu to 28Si, 29Si i 30Si. Wartościowość w większości związków wynosi 4, rzadziej spotykane są związki z krzemem dwuwartościowym. Typowe stopnie utlenienia to –IV i IV, rzadko –II i II; znane są też związki o st. utl. 0. Krzem (w postaci monokryształów) jest wykorzystywany powszechnie w przemyśle elektronicznym.




Metoda flux-melt (inaczej krystalizacja z roztworu bezwodnego) - metoda otrzymywania monokryształów polegająca na stosowaniu topników tworzących roztwór z krystalizowanym materiałem.
W metodzie tej krystalizowaną mieszaninę podgrzewa się w platynowym tyglu stale mieszając, aż do uzyskania jednorodnego, bezwodnego roztworu. Powolne chłodzenie, które może trwać wiele dni, prowadzi do wykrystalizowania z roztworu monokryształów. Cały proces przebiega w warunkach normalnego ciśnienia. Metodą tą można wytwarzać kryształy raczej niskiej jakości, o ograniczonej jednorodności i czystości.
Krzem został zidentyfikowany jako pierwiastek przez Antoine Lavoisiera w 1787. Humphry Davy, w 1800 r. błędnie uznał, że krzem jest związkiem chemicznym i opinia ta przetrwała aż do 1824 r., kiedy to Jöns Jacob Berzelius otrzymał czysty krzem z krzemionki SiO2, przeprowadzając ją kwasem fluorowodorowym w SiF4i redukując go potasem.


(...) Występowanie krzemu: Zawartość krzemu w zewnętrznych strefach Ziemi wynosi 26,95% wagowo. Jest drugim po tlenie najbardziej rozpowszechnionym pierwiastkiem. Krzemionka SiO2 w różnych odmianach polimorficznych (kwarc, trydymit, krystobalit) oraz krzemiany i glinokrzemiany stanowią większość skał tworzących skorupę ziemską. Od niego pochodzi nazwa pierwszej, zewnętrznej warstwy globu SiAl.
Przeciętna zawartość krzemu w glebie jest podobna jak w litosferze, ale w zależności od typu może być niższa niż 1% lub bliska 50%. Zawartość w wodach podziemnych jest różna, w porównaniu z zawartością w skałach niska, gdyż związki krzemu są słabo rozpuszczalne, choć wystarczająca do uznania krzemu za makroelement. Największe stężenie rozpuszczonych związków krzemu występuje w wodach termalnych, gdzie przekracza 100, a nawet 600 mg/dm³, jednak średnia zawartość krzemionki rozpuszczonej w wodach podziemnych to kilkanaście mg/dm³. Zawartość w tkankach roślinnych mieści się w zakresie od kliku setnych procenta suchej masy (np. w korzeniach buraka cukrowego), przez kilka dziesiętnych procenta (np. w pędach roślin motylkowatych), kilka procent (wiele zbóż), kilkanaście procent (np. w skrzypach), do 20% (w ryżu). W uproszczeniu w tkankach roślin jednoliściennych jest o rząd wielkości wyższa niż w dwuliściennych. Pewne ilości krzemu znajdują się też w organizmach zwierząt.
Związki: Krzem, podobnie jak węgiel, tworzy łańcuchy krzem-krzem, krzem-tlen-krzem oraz krzem-azot-krzem. Istnieje dość liczna (około 300 000) grupa takich związków, jest ich jednak o wiele mniej niż związków węgla. Ze względu na zdolność do tworzenia łańcuchów, krzem jest proponowany jako alternatywna wobec węgla podstawa życia. Najważniejsze związki krzemu to krzemionka, będąca podstawowym składnikiem piasku i szkła, kwasy krzemowe H2nSimO2m+n, ich sole – krzemiany, które są składnikami szkła wodnego oraz chlorosilany i alkoksysilany podstawowe substraty do produkcji polisiloksanówi żeli krzemionkowych.
Znaczenie biologiczne krzemu: Organizm ludzki potrzebuje 20-30 mg krzemu dziennie. Większej dawki wymagają kobiety w ciąży, osoby po operacjach kostnych oraz ludzie starsi, gdyż ilość tego pierwiastka w narządach maleje z wiekiem. Występuje przede wszystkim w tkance łącznej, z której zbudowane są ścięgna, błony śluzowe, ściany naczyń krwionośnych, zastawki serca, skóra i układ kostno-stawowy. Krzem usuwa z komórek substancje toksyczne, korzystnie wpływa na naczynia włosowate, uszczelniając je, zwiększa wytrzymałość tkanki kostnej, wzmacnia zdolność obronną organizmu przeciw zakażeniom, zapobiega przedwczesnemu starzeniu się. Usuwa podrażnienia i stany zapalne skóry, poprawiając jej ogólny wygląd i zapobiegając wiotczeniu, ogranicza wypadanie włosów, przyspiesza ich wzrost, wzmacnia paznokcie. Jest on również używany do budowy ścian komórkowych niektórych organizmów oraz stanowi centrum reaktywności kilkunastu enzymów, odpowiedzialnych za „przerób” krzemionki okrzemek i niektórych skorupiaków. Krzem występuje w wielu roślinach, którym jest potrzebny do prawidłowego rozwoju, jednak nie udało się udowodnić, aby był niezbędny do rozwoju wszystkich gatunków. Zwykle jego obecność zwiększa odporność na agrofagi, zwłaszcza grzyby, które mają utrudnione wnikanie w tkanki roślin wysycone krzemionką. Podobnie w przypadku zwierząt, niezbędność krzemu wykazano dla gąbek krzemionkowych, ale mimo że występuje w ciałach wszystkich zwierząt, zwykle nie udowodniono dla nich jego niezbędności. U kręgowców występuje w większych ilościach we włosach i piórach (np. wełna owiec zawiera 0,02–0,08% SiO2). Krzem jest niezbędny dla rozwoju okrzemek i wykracza to poza rolę budulca ich skorupek.

*Krzem amorficzny zwany a-Si - to niekrystaliczny alotrop pozyskiwany z krzemu, tzw. krzem w fazie amorficznej, masowo wykorzystywany przy produkcji ogniw fotowoltaicznych[1], wyświetlaczy LCD, OLED. Żywotność krzemu w postaci bezkształtnej jest ponad dwukrotnie niższa od krzemu monokrystalicznego i wynosi ok. 10 lat.





Związki krzemu:
a) Nieorganiczne związki krzemu:






--> Krzemiany (Minerały):


Lista krzemianów: KLIK!.
b) Organiczne związki krzemu:

















 3) German (Ge, łac. Germanium) - pierwiastek chemiczny z bloku p układu okresowego. Jest twardym, błyszczącym, srebrzystoszarym półmetalem o właściwościach chemicznych podobnych do innych węglowców, przede wszystkim krzemu i cyny. Tworzy wiele związków organicznych.
3) German (Ge, łac. Germanium) - pierwiastek chemiczny z bloku p układu okresowego. Jest twardym, błyszczącym, srebrzystoszarym półmetalem o właściwościach chemicznych podobnych do innych węglowców, przede wszystkim krzemu i cyny. Tworzy wiele związków organicznych.
Odkrycie: Mimo że występuje w skorupie ziemskiej w stosunkowo dużych ilościach, został odkryty późno, ponieważ niewiele minerałów zawiera go w dużym stężeniu. W 1869 roku Dymitr Mendelejewprzewidział jego istnienie i niektóre właściwości na podstawie luki, jaka istniała w jego układzie okresowym (podobnie przewidział istnienie skandu i galu). Nazwał go ekakrzemem. Prawie 20 lat później, w 1886 roku, Clemens Winkler odkrył nowy pierwiastek, badając minerał argyrodyt, i nadał mu nazwę german (od łacińskiej nazwy swojego kraju ojczystego – Niemiec). Wyniki obserwacji Winklera potwierdziły przypuszczenia Mendelejewa. German posiada kilkanaście izotopów z przedziału mas 64-83, z czego 5 trwałych występuje w naturalnej mieszaninie.
Zastosowania: German jest ważnym półprzewodnikiem, wykorzystywanym do produkcji tranzystorów, diod i innych elementów elektronicznych. Jego pasmo wzbronione ma szerokość 0,67 eV, jest więc węższe niż w przypadku krzemu. Podobnie jak w przypadku galu, sole germanu – zwłaszcza fluorki i arsenki – wykazują własności półprzewodnikowe i elektroluminescencyjne, jednak ze względu na większą dostępność galu, związki te nie są praktycznie wykorzystywane. German jest również stosowany do produkcji światłowodów i katalizatorów polimeryzacji. Pozyskuje się go głównie z zanieczyszczeń w minerale sfalerycie, a także z zanieczyszczeń rud cynku, ołowiu, miedzi i srebra.
Reaktywność: Nie reaguje z wodą, powietrzem, a nawet z kwasami i zasadami, oprócz kwasu azotowego. Czasami domieszkuje się nim krzem stosowany przy produkcji elementów elektronicznych, jednak ze względu na małą dostępność tego pierwiastka zwykle jest on zastępowany galem. Szkłodomieszkowane germanem jest przezroczyste dla promieniowania podczerwonego. Jego znaczenie biologiczne jest nieznane; niektóre związki germanu, takie jak germanowodór czy chlorek germanu, mają działanie drażniące.
Odmiany alotropowe: W 2014 dwa zespoły naukowców, chiński i europejski otrzymały germanen – materiał tworzony przez płaską strukturę atomów germanu, o budowie analogicznej do grafenu i silicenu.


Atomy germanu tworzą w nim jednoatomowej grubości warstwę, połączone są w sześciokąty na podobieństwo plastra miodu.


 4) Cyna (Sn, łac. stannum) - jest to pierwiastek chemiczny, metal z bloku p w układzie okresowym. Cyna tworzy odmiany alotropowe. W warunkach standardowych występuje w odmianie β (beta), zwanej cyną białą, trwałej powyżej 13,2 °C. Odmiana ta ma sieć krystaliczną w układzie tetragonalnym, o gęstości 7,3 g/cm³. W niższej temperaturze przechodzi w odmianę regularną α (alfa) o gęstości 5,85 g/cm³. Zmiana gęstości jest równoznaczna ze zmianą objętości, co powoduje, że cyna rozpada się, tworząc szary proszek zwany cyną szarą. Zjawisko to nazywane jest zarazą cynową. Czysta cyna biała jest ciągliwa i kowalna, odporna na korozję.
4) Cyna (Sn, łac. stannum) - jest to pierwiastek chemiczny, metal z bloku p w układzie okresowym. Cyna tworzy odmiany alotropowe. W warunkach standardowych występuje w odmianie β (beta), zwanej cyną białą, trwałej powyżej 13,2 °C. Odmiana ta ma sieć krystaliczną w układzie tetragonalnym, o gęstości 7,3 g/cm³. W niższej temperaturze przechodzi w odmianę regularną α (alfa) o gęstości 5,85 g/cm³. Zmiana gęstości jest równoznaczna ze zmianą objętości, co powoduje, że cyna rozpada się, tworząc szary proszek zwany cyną szarą. Zjawisko to nazywane jest zarazą cynową. Czysta cyna biała jest ciągliwa i kowalna, odporna na korozję.
Cynowa zaraza - zjawisko przemiany alotropowej cyny białej w szarą i odwrotnie.
Zastosowania cyny: Ze względu na dostępność, niską temperaturę topnienia, łatwość odlewania oraz dobre własności mechaniczne przedmioty cynowe były bardzo popularne od wczesnego średniowiecza; największy rozkwit produkcji przedmiotów z cyny (konwisarstwa) miał miejsce pomiędzy XIV i XVI w.

(...) Cyny używa się do pokrywania innych metali cienką warstwą antykorozyjną. Proces cynowania stosowany jest do zabezpieczania naczyń stalowych, stosowanych w przemyśle spożywczym, np. puszek do konserw oraz konwi na mleko.

Stop cyny i ołowiu ma niską temperaturę topnienia (np. przy 61,9% wag. cyny jest to temperatura 183 °C) i stosowany był w przemyśle i elektrotechnice jako lut do łączenia innych metali poprzez lutowanie. Po 1 lipca 2006 w związku z wycofywaniem w krajach Unii Europejskiej produktów zawierających ołów przechodzi się na lutowanie bezołowiowe, zastępując ołów niewielkim dodatkiem srebra, miedzi i bizmutu.





(...) Cyna jest również składnikiem stopu drukarskiego do wyrobu czcionek Występuje w nim wraz z ołowiem (głównym składnikiem) oraz antymonem. Stopy drukarskie występują w różnych odmianach (charakteryzujących się różną twardością) różniących się stosunkiem składników.



Stopami cyny i miedzi są brąz cynowy oraz spiż (zawierający także cynk i ołów), używany do odlewania dzwonów.


Występowanie: Najczęściej spotykane minerały zawierające cynę to tlenek – kasyteryt (SnO2, 78,8% Sn) i siarczek – stannin (Cu2FeSnS4, 27,6% Sn). Cyna występuje w przyrodzie w ilości 0,004% wagowo. Największe złoża cyny mieszczą się w Indonezji, Chinach, Tajlandii, Boliwii, Malezji, Rosji, Brazylii, Birmie i Australii. W Polsce większe złoża cynonośne występowały w Gierczynie i stanowiły jedyne poważniejsze źródło cyny w Polsce. W ilościach śladowych cyna występuje także w Sudetach, w okolicach Czarnowa (arsenopiryt), Miedzianki, i Starej Góry.
Izotopy: Cyna posiada 10 występujących w przyrodzie trwałych izotopów. Najbardziej rozpowszechniony jest izotop 120Sn (ok. 33%). Sztucznie wytworzono dalszych 29 izotopów cyny o masie atomowej od 99 do 137 i okresie połowicznego rozpadu od kilku ms do 230 tys. lat.
*Babbit – stop łożyskowy z zawartością 83–88% cyny, 8–10% antymonu, 3–6% miedzi i 0,5% ołowiu. Stop ten jest używany na silnie obciążonych panewkach łożysk ślizgowych, np. w silnikach lotniczych i samochodowych. Opracowany w 1839 roku przez Isaaca Babbitta w Taunton, Massachusetts, USA.
*Britannia (metal britannia, początkowo także biały metal) – stop metali zawierający 92% cyny, 6% antymonu i 2% miedzi, po raz pierwszy uzyskany w Sheffield w drugiej połowie XVIII wieku.



*Nordic gold (GN, NG, golden nordic, nordyckie złoto, CuAl5Zn5Sn1) – jest to stop 89% miedzi (Cu), 5% cynku (Zn), 5% aluminium (Al) oraz 1% cyny (Sn). Gold (Złoto) w nazwie nie oznacza, że ten stop zawiera chociaż śladową ilość złota, jest to tylko określenie koloru. Od prawdziwego złota łatwo go odróżnić ze względu na znacznie mniejszą gęstość. W Polsce używany do produkcji monet okolicznościowych (obiegowych) o nominale 2 zł. Z tego stopu są również wybijane eurocenty (10, 20, 50) oraz 10-koronówki w Szwecji. Bez odpowiedniego zabezpieczenia (np. kapsla ochronnego) stop ten łatwo koroduje na powietrzu tracąc swój pierwotny blask. Monety wykonane ze złota nordyckiego pokrywają się bardzo szybko patyną, a najlepszym sposobem ich przechowywania w zbiorach numizmatycznych są specjalne pudełka próżniowe (tzw. slaby numizmatyczne).

*Stop Lichtenberga – niskotopliwy stop metali o składzie wagowym:
- bizmut 50%
- ołów 30%
- cyna 20%
Jego temperatura topnienia określana jest jako 91,6 °C lub 100 °C. Został otrzymany przez niemieckiego naukowca Georga Christopha Lichtenberga.









*Stop Lipowitza – niskotopliwy stop metali o składzie wagowym:
- bizmut 50%
- ołów 27%
- cyna 13%
- kadm 10%
Jego temperatura topnienia wynosi 70–73 °C.
Stosowany jest w radioterapii do wykonywania osłon ochronnych przed promieniowaniem.
*Stop Newtona – niskotopliwy stop metali o składzie wagowym:
- bizmut 50%
- ołów 31,2%
- cyna 18,8%
Jego temperatura topnienia wynosi 96–97 °C[1][3].
Zastosowania: dentystyka, modelarstwo, mechanika i odlewnictwo precyzyjne. Jest bezpieczniejszą (nie zawiera kadmu) alternatywą dla stopu Lipowitza do wykonywania osłon przed promieniowaniem podczas radioterapii.
Odkrywcą tego stopu jest Isaac Newton.
Znany jest też inny niskotopliwy stop określany mianem stopu Newtona, który zawiera ołów, bizmut i kadm w stosunku wagowym 7:4:1 i topi się on w temperaturze 95 °C.



*Zircaloy - stop cyrkonu na koszulki elementów paliwowych w reaktorach jądrowych. Zircaloy oprócz cyrkonu zawiera ok. 1,5–2,5% cyny oraz niewielkie dodatki niklu, chromu, niobu i żelaza. Zaletą zircaloy jest to, iż wytrzymuje warunki panujące w rdzeniu reaktora i jest prawie przezroczysty dla neutronów termicznych (tzn. ma dla nich bardzo mały przekrój czynny).
ZWIĄZKI CYNY:








 5) Ołów - pierwiastek chemiczny o symbolu Pb(z łaciny plumbum) i liczbie atomowej 82. Należy do metali ciężkich, jest gęstszy od większości popularnych materiałów. Miękki i kowalny, ma też względnie niską temperaturę topnienia. Świeżo cięty jest białawo-niebieski, na powietrzu matowieje do matowej szarości. Ma najwyższą liczbę atomową ze wszystkich stabilnych pierwiastków, kończy trzy szeregi rozpadu cięższych pierwiastków promieniotwórczych.
5) Ołów - pierwiastek chemiczny o symbolu Pb(z łaciny plumbum) i liczbie atomowej 82. Należy do metali ciężkich, jest gęstszy od większości popularnych materiałów. Miękki i kowalny, ma też względnie niską temperaturę topnienia. Świeżo cięty jest białawo-niebieski, na powietrzu matowieje do matowej szarości. Ma najwyższą liczbę atomową ze wszystkich stabilnych pierwiastków, kończy trzy szeregi rozpadu cięższych pierwiastków promieniotwórczych.
Jest względnie niereaktywnym metalem grup głównych. Jego słaby metaliczny charakter ilustrują właściwości amfoteryczne; ołów i tlenki ołowiu reagują z kwasami i zasadami, ma on tendencję do tworzenia wiązań kowalencyjnych. Związki chemiczne zazwyczaj zawierają ołów na stopniu utlenienia raczej +2 niżeli +4, częstszym u lżejszych przedstawicieli węglowców. Wyjątki ograniczają się głównie do związków organicznych. Może tworzyć łańcuchy, pierścienie i struktury wielościenne.
Ołów łatwo otrzymać z rudy, wiedziano o tym już w prehistorycznej Azji Zachodniej. Główna ruda ołowiu, galena, często zawiera srebro, zainteresowanie którym pomogło zapoczątkować szeroko rozpowszechnione wydobycie i użytkowanie ołowiu w Starożytnym Rzymie. Produkcja ołowiu spadła po upadku Cesarstwa, nie osiągając porównywalnych poziomów aż do rewolucji przemysłowej. W 2014 globalna roczna produkcja wyniosła około 10 milionów ton, z czego ponad połowa pochodziła z recyklingu. Wysoka gęstość ołowiu, niska temperatura topnienia, przewodność i względnie mała podatność na utlenianie stanowią o jego użyteczności. W połączeniu z jego względną obfitością i niskimi kosztami skutkowały jego powszechnym użyciem w budowlach, hydraulice, bateriach, nabojach i śrucie, wagach, lutownictwie, pewterze i innych stopach, białych farbach, benzynie i osłonach przed promieniowaniem.
Ołów łatwo otrzymać z rudy, wiedziano o tym już w prehistorycznej Azji Zachodniej. Główna ruda ołowiu, galena, często zawiera srebro, zainteresowanie którym pomogło zapoczątkować szeroko rozpowszechnione wydobycie i użytkowanie ołowiu w Starożytnym Rzymie. Produkcja ołowiu spadła po upadku Cesarstwa, nie osiągając porównywalnych poziomów aż do rewolucji przemysłowej. W 2014 globalna roczna produkcja wyniosła około 10 milionów ton, z czego ponad połowa pochodziła z recyklingu. Wysoka gęstość ołowiu, niska temperatura topnienia, przewodność i względnie mała podatność na utlenianie stanowią o jego użyteczności. W połączeniu z jego względną obfitością i niskimi kosztami skutkowały jego powszechnym użyciem w budowlach, hydraulice, bateriach, nabojach i śrucie, wagach, lutownictwie, pewterze i innych stopach, białych farbach, benzynie i osłonach przed promieniowaniem.
Pod koniec XIX wieku odkryto zatrucie ołowiem. Od tego czasu użycie ołowiu wycofano z wielu zastosowań. Ołów stanowi neurotoksynę, która akumuluje się w tkankach miękkich i kościach, uszkadza układ nerwowy i krew. Szczególnie szkodzi dzieciom: nawet w przypadku znormalizowanego w wyniku leczenia stężenia we krwi może dojść do trwałego uszkodzenia mózgu.
Właściwości fizyczne:
-> Atomowe: Atom ołowiu liczy 82 elektrony o konfiguracji [Xe]4f145d106s26p2. Połączone pierwsza i druga energia jonizacji — całkowita energia potrzebna do usunięcia dwóch elektronów z orbitalu 6p — leży blisko analogicznej wartości dla cyny, węglowca leżącego w układzie okresowym pierwiastków nad ołowiem. Nie jest to częste zjawisko, zazwyczaj energia jonizacja spada wraz ze schodzeniem w dół danej grupy, kiedy to zewnętrzne elektrony kolejnych pierwiastków leżą coraz dalej od jądra atomowego i są coraz bardziej ekranowane przez bliższe orbitale. Podobieństwo energii jonizacji spowodowane jest kontrakcją lantanowców – spadkiem promienia atomu od lantanu (o liczbie atomowej 57) do lutetu (71) – i względnie małymi promieniami pierwiastków po hafnie (72). Wynika to ze słabego ekranowania jądra przez elektrony 4f lantanowców. Dodane pierwsze cztery energie jonizacji ołowiu przekraczają rzeczoną wartość dla cyny, co stoi w sprzeczności do przewidywań na podstawie układu okresowego. Do tego efektu przyczyniają się efekty relatywistyczne, odgrywające znaczniejszą rolę w cięższych pierwiastkach. Około 10% kontrakcji lantanowców przypisano takim właśnie efektom. Należy do nich inert pair effect: elektrony 6s ołowiu niechętnie tworzą wiązania chemiczne, co czyni odległości pomiędzy najbliższymi sobie atomami w sieci krystalicznej niezwykle długimi.

















-> Atomowe: Atom ołowiu liczy 82 elektrony o konfiguracji [Xe]4f145d106s26p2. Połączone pierwsza i druga energia jonizacji — całkowita energia potrzebna do usunięcia dwóch elektronów z orbitalu 6p — leży blisko analogicznej wartości dla cyny, węglowca leżącego w układzie okresowym pierwiastków nad ołowiem. Nie jest to częste zjawisko, zazwyczaj energia jonizacja spada wraz ze schodzeniem w dół danej grupy, kiedy to zewnętrzne elektrony kolejnych pierwiastków leżą coraz dalej od jądra atomowego i są coraz bardziej ekranowane przez bliższe orbitale. Podobieństwo energii jonizacji spowodowane jest kontrakcją lantanowców – spadkiem promienia atomu od lantanu (o liczbie atomowej 57) do lutetu (71) – i względnie małymi promieniami pierwiastków po hafnie (72). Wynika to ze słabego ekranowania jądra przez elektrony 4f lantanowców. Dodane pierwsze cztery energie jonizacji ołowiu przekraczają rzeczoną wartość dla cyny, co stoi w sprzeczności do przewidywań na podstawie układu okresowego. Do tego efektu przyczyniają się efekty relatywistyczne, odgrywające znaczniejszą rolę w cięższych pierwiastkach. Około 10% kontrakcji lantanowców przypisano takim właśnie efektom. Należy do nich inert pair effect: elektrony 6s ołowiu niechętnie tworzą wiązania chemiczne, co czyni odległości pomiędzy najbliższymi sobie atomami w sieci krystalicznej niezwykle długimi.
Lżejsi od ołowiu członkowie jego grupy tworzą stabilne bądź metastabilne odmiany alotropowe o tetraedrycznej powiązanej kowalencyjnie strukturze przestrzennej diamentu. Poziomy energetyczne ich zewnętrznych orbitali s i p leżą wystarczająco blisko, by umożliwić hybrydyzację do orbitali sp3. W przypadku ołowiu inert pair effect zwiększa rozdział między orbitalami s i p. Przerwa między nimi nie może zostać przezwyciężona przez energię, która wydzieli się przez tworzenie dodatkowych wiązań dzięki hybrydyzacji. Raczej niż przestrzenna struktura diamentu, w ołowiu wytwarzają się wiązania metaliczne, w których jedynie zdelokalizowane elektrony p są dzielone między jonami Pb2+. W efekcie ołów przyjmuje układ regularny, jak podobnej wielkości dwuwartościowe metalu wapń czy stront. Tetraedryczna forma alotropowa cyny zwie się cyną α lub szarą i jest stabilna jedynie w temperaturze ≤ 13,2 °C. Powyżej tej wartości stabilną jest forma β, biała cyna o zniekształconej strukturze układu regularnego, która można wyprowadzić poprzez zgniecenie czworościanu cyny szarej wzdłuż osi sześciennych. Cyna biała ma efektywnie strukturę pośrednią między tetraedryczną strukturą cyny szarej i regularną strukturą ołowiu, zgodnie z ogólnym trendem zwiększania się własności metalicznych w dół grupy. Kwazikrystaliczna cienkowarstwowa forma alotropowa ołowiu o symetrii pentagonalnej odkryta została w 2013. Uzyskano ją przez odkładanie atomów ołowiu na powierzchni ikozaedrycznego kwazikryształu srebra, indu i iterbu. Nie odnotowano przewodnictwa tej postaci.


Izotopy ołowiu: Naturalny ołów składa się z czterech stabilnych izotopów o liczbach masowych 204, 206, 207 i 208 oraz śladowych ilości pięciu izotopów o krótkim okresie półtrwania. Wysoka liczba izotopów zgadza się z parzystą liczbą atomową ołowiu (parzysta liczba protonów lub neutronów zwiększa stabilność jądra, żaden pierwiastek o nieparzystej liczbie atomowej nie posiada więcej niż dwóch stabilnych izotopów, te o parzystych liczbach mają zaś wiele stabilnych izotopów, np cyna o liczbie atomowej 50 posiada ich najwięcej, aż 10). Ołów ma magiczną liczbę protonów (82), dla której model powłokowy jądra atomowego trafnie przewiduje szczególną stabilność jądra. Izotop 208 ma 126 neutronów, co może wyjaśniać, czemu jest niezwykle stabilny.















(...) Ze swą wysoką liczbą atomową, ołów jest najcięższym pierwiastkiem, którego naturalne izotopy uznaje się za stabilne. Izotop 208 ma najcięższe stabilne jądro. Tytuł ten poprzednio przysługiwał bizmutowi o liczbie atomowej 83, nim jego jedyny wykryto powolny rozpad jego jedynego uznawanego za stabilny izotopu 209 w 2003. Określony eksperymentalnie okres połowicznego zaniku wyniósł 1,9×1019 lat. Kilogram naturalnego bizmutu miałby więc aktywność szacunkowo 0,003 bekereli (rozpadów na s), podczas gdy aktywność naturalnego promieniowania w ciele ludzkim wynosi około 65 Bq/kg. 4 stabilne izotopy ołowiu mogą teoretycznie przechodzić rozpad alfa do izotopów rtęci z uwolnieniem energii, jednak dotychczas przemian takich nie zaobserwowano. Przewidywane okresy półtrwania mieszczą się w zakresie od 1035 do 10189 lat.
3 ze stabilnych izotopów leżą w czterech głównych łańcuchach rozpadu: 206, 207 i 208 są izotopami kończącymi rozpad uranu 238, uranu 235 i toru 232. Łańcuchy te określa się serią uranową, aktynową i torową. Stężenie izotopów w skałach naturalnych zależy w dużym stopniu od obecności wspomnianych izotopów uranu i toru. Przykładowo względna obfitość ołowiu 208 może sięgać od 52% w zwykłych próbkach do 90% w rudach toru. Z tej przyczyny standardowa masa atomowa ołowiu podawana jest z tylko jednym miejscem po przecinku. Z biegiem czasu iloczyn ilości izotopów 206 i 207 w stosunku do 204 narasta, jako że dwa pierwsze są suplementowane przez rozpada radioaktywny cięższych pierwiastków, w przeciwieństwie do ołowiu 204. Pozwala to na datowanie metodą ołów-ołów. Gdy uran rozpada się do ołowiu, względna ilość tego ostatniego rośnie. Stanowi to z kolei podstawę metody datowania uran-ołów.
Poza stabilnymi izotopami, z których składa się prawie cały występujący naturalnie ołów, istnieją również śladowe ilości kilku izotopów promieniotwórczych. Należy do nich ołów 210; chociaż jego okres półtrwania wynosi jedyni 22,3 roku, niewielkie jego ilości występują w naturze, gdyż powstaje on w długiej serii rozpadów rozpoczynającej się uranem 238, obecnym na Ziemi od miliardów lat. Izotopy 211, 212 i 214 obecne są w łańcuchach odpowiednio rozpadu uranu 235, toru 232 i uranu 238, wobec czego śladowe ilości wszystkich trzech występują naturalnie. Niewielkie ślady ołowiu 209 powstają z bardzo rzadkiego rozpadu klasterowego radu 223, którego jednym z produktów jest naturalny uran 235. Ołów 210 jest szczególnie użyteczny w identyfikacji wieku próbek poprzez pomiar jego stosunku do ołowiu 206 (oba izotopy występują w jednym łańcuchu rozpadu).


 |
| Źródło: Wikipedia. Meteoryt Holsinger, najwięk- -szy kawał meteorytu Canyon Diablo. Datowanie metodą uran-ołów i ołów-ołów pozwoliło na dokładniejsze określenie wieku Ziemi na 4,55 miliarda +/- 70 milionów lat. |
(...) W sumie zsyntetyzowano 43 izotopy ołowiu o liczbach masowych 178–220. Najstabilniejszym z nich jest ołów 205 o okresie połowicznego rozpadu 15 milionów lat. Rozpada się on wyłącznie przez wychwyt elektronu, co oznacza, że kiedy nie ma dostępnych żadnych elektronów i atom jest w pełni zjonizowany (jądro bez żadnego z 82 elektronów), nie może się rozpaść. W pełni zjonizowany tal 205, do którego rozpada się ołów 205, nie jest stabilny i może rozpadać się dalej do ołowiu 205. Drugi najbardziej stabilny jest ołów 202 o czasie połowicznego zaniku 53000 lat, a więc dłuższym, niż ma którykolwiek z naturalnych śladowych radioizotopów.



















Ograniczenia i rekultywacja:

Do połowy lat 80. XX wieku nastąpiła znacząca zmiana użytkowania ołowiu. W USA regulacje środowiskowe zredukowały bądź wyeliminowały w ogóle wykorzystanie ołowiu w produktach innych od baterii, wliczając w to benzynę, farby, lut i systemy wodne. Elektrownie czerpiące energię ze spalania węgla stosować mogą różne urządzenia wychwytujące emitowany ołów. Dalej użytkowanie ołowiu ogranicza unijna dyrektywa RoHS. Używania śrutu ołowianego w polowaniach i strzelectwie sportowym zabroniła Holandia w 1993, wywołując tym samym znaczący spadek emisji ołowiu z 230 ton w 1990 do 47,5 tony w 1995[246].
W USA istnieje granica dopuszczalnej ekspozcji na ołów w miejscu pracy, obejmuje ona ołów metaliczny, nieorganiczne związki ołowiu i mydła ołowiu, a wynosi 0,05 mg/m3 dla 8-godzinnego dnia pracy. Zalecane stężenie ołowiu we krwi ma górną granicę 0,04 mg na 100 g masy ciała. Ołów może być wciąż obecny w szkodliwych ilościach w wyrobach kamionkowych, winylowych (jak te używane do produkcji rur i ozilacji przewodów elektrycznych) w chińskim mosiądzu, w istocie stopie miedzi i cynku z dodatkiem ołowiu, żelaza, cyny i czasami antymonu. Stare domy mogą być wciąż pokryte ołowianymi farbami. Biała farba ołowiana została wycofana ze sprzedaży w krajach uprzemysłowionych, utrzymały się jednak specjalistyczne zastosowania innych pigmentów, na przykład żółtego chromianu (VI) ołowiu (II). Zdrapywanie starej farby podczas szlifowania produkuje pył, które może zostać wdechnięty[250]. Programy redukcji ołowiu zostały wprowadzone przez niektóre władze w nieruchomościach związanych z małymi dziećmi[251].
Odpady zawierające ołów, w zależności od jurysdykcji i natury samych odpadów, można traktować jako odpady komunalne (co ułatwia złagodzenie wymagań utylizacji) bądź jako potencjalnie niebezpieczne odpady wymagające specjalnego obchodzenia się i składowania. Przeprowadzono badania dotyczące usuwania ołowiu z układów biologicznych za pomocą metod biologicznych. Kości rybie posiadają zdolność bioremediacji ołowiu ze skażonej gleby. Kropidlak różnobarwny jest efektywny w usuwanie jonów ołowiu. Przebadano zdolności kilku bakterii do usuwania ołowiu ze środowiska, wliczając w to bakterie redukujące siarczany Desulfovibrio i Desulfotomaculum, oba rodzaje wysoce efektywne w roztworach wodnych.





*Rafinacja ołowiu - jest to proces oczyszczania surowego ołowiu (otrzymywanego w procesach IS, QSL, w piecu Dorschla lub w piecu Kaldo) z zanieczyszczeń. Surowy ołów (ok. 99% Pb) po procesie rafinacji powinien zawierać:
- PB990R – 99,990% Pb
- PB985R – 99,985% Pb
- PB970R – 99,970% Pb
- PB940R – 99,940% Pb
Szlikrowanie i odmiedziowanie likwacyjne: Operacje szlikrowania i odmiedziowania likwacyjnego przeprowadza się w tych samych kotłach, w których ołówzostał roztopiony. Proces ten rozpoczyna się od podgrzania ołowiu do temperatury ok. 450 °C. Przekroczenie tej temperatury powoduje opóźnienie procesu rafinacji oraz nadmierneme zużycie gazu. Po osiągnięciu zadanej temperatury należy przerwać opalanie i wstawić mieszadło szybkoobrotowe, celem wymieszania całej zawartości kotła. W wyniku mieszania następuje wydzielenie się szlikrów, które wypływają na powierzchnię kąpieli. Czas mieszania kąpieli zależy od ilości zanieczyszczeń, które przejdą do szlikrów. Większa ilość zanieczyszczeń przedłuża czas mieszania. Operację szlikrowania należy tak prowadzić, aby szlikry zawierały jak najmniejszą ilość ołowiu. W celu oddzielenia szlikirów od ołowiu dodaje się zwilżone trociny drzewne do leja wytworzonego przez szybko obrotowe mieszadło. Czas mieszania wynosi około 45-60min. W wyniku mieszania szlikry otrzymują postać pylistą. Powstałe szlikry należy zdjąć z powierzchni ołowiu. Po całkowitym oczyszczeniu powierzchni kąpieli należy pobrać próbę ołowiu dla określenia składu chemicznego. Uzyskany wynik analityczny jest podstawą do postępowania technologicznego w następnych operacjach rafinacyjnych. Analiza powinna podawać zawartość: Ag, Sb, As, Sn, Cu, Bi, Zn, Tl, In, Cd i Fe. Dla uzyskania właściwego wypełnienia kotła należy poziom uzupełnić surowym ołowiem. Po zakończeniu operacji szklirowania należy przystąpić do procesu likwacyjnegoodmiedziowania. W wyniku obniżenia temperatury kąpieli z ołowiu wypływają kryształy miedzi oraz związki międzymetaliczne miedzi z arsenem, cyną i antymonem, które krzepną na powierzchni ołowiu. W temperaturze około 330 °C należy ołów przepompować do następnego kotła przeznaczonego do procesu odmiedziowania siarką. Zakończenie pompowania i zdemontowanie układu pompowania kończy proces szlikrowania i odmiedziowania likwacyjnego.
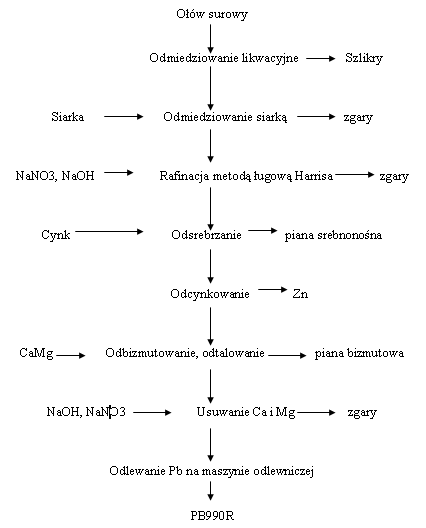 |
| Źródło: Wikipedia. Schemat procesu rafinacji ołowiu. |
- siarkę elementarną granulowaną
- galenę z wysoką zawartością PbS
- Piryt o dużej zawartości FeS2
Z uwagi na największą dostępność stosuje się mieszankę S i PbS w celu ograniczenia upału siarki w czasie jej wsadowania do kotła oraz dla przyśpieszenia procesu. W czasie odmiedziowania siarką zachodzą następujące reakcje:
- Pb + S = PbS
- PbS + Cu = CuS + Pb
- Chemicznie czysty PbS wnosi 13,3% S
- Chemicznie czysty FeS wnosi 33,4% S
- Chemicznie czysty FeS2 wnosi 53,3% S
- Jeżeli zawartość miedzi wynosi powyżej 0,005% to operację odmiedziowania siarką należy powtórzyć.
- Rafinacja metodą Harrisa, usuwanie Sn, As, Sb, Zn: Ołów po odmiedziowaniu zawiera jeszcze inne zanieczyszczenia takie jak: Sn, As, Sb, Bi, Ag, Au oraz sodę kaustyczną NaOH, która pełni rolę kolektora dla powstałych tlenków. Wstępna rafinacja ma na celu usunięcie arsenu, cyny i antymonu do takich wartości, aby osiągnąć sprzyjające warunki dla późniejszego odzysku srebra. Proces ten jest selektywny. Utlenianie domieszek następuje w kolejności: As, Sn i Sb. Uważa się, że w obecności NaOH i NaNO3 zachodzą następujące reakcje:
- 5Pb + 2NaNO3 = Na2O + 5PbO + N2
- PbO + Na2O = Na2PbO2
- 4PbO + 8NaOH = 4Na2PbO2 + 4H2O
- 2As + 5Na2PbO2 + 2H2O = 2Na3AsO4 + 4NaOH + 5Pb
- Sn + 2Na2PbO2 + H2O = Na2SnO3+ 2NaOH + 2Pb
- 2Sb + 5Na2PbO2 + 2H2O = 2Na3SbO4 + 4NaOH + 5Pb
Temperatura procesu wynosi ok. 480-500 °C, czas mieszania wynosi ok. 180min. Pod koniec procesu należy zebrać zgary. Rafinację wstępną uważa się za zakończoną, jeżeli zawartość zanieczyszczeń wynosi maksymalnie- As – 0,001%
- Sn – 0,001%
- Sb – 0,2%
- *Zgar – strata metalu w czasie procesów metalurgicznych głównie wytapiania, jest wynikiem reakcji chemicznej, najczęściej utlenianiametalu i pierwiastków w nim zawartych. Produktem takiej reakcji są głównie tlenki występujące w stanie stałym i przechodzą do żużla. Niekiedy zgary występują w formie gazowej (np. CO) . Zgary są źródłem strat otrzymywanego pierwiastka.
- Odrebrzanie: Ołów oczyszczony z Cu, Sn, As i Sb zawiera jeszcze Ag. Proces odsrebrzania Pb prowadzi się metodą Parkesa. Polega on na dodawaniu Zn do płynnego ołowiu. Odsrebrzanie ołowiu przeprowadzać należy w ten sposób, aby:
- czas operacji był jak najkrótszy
- otrzymywać jak najmniejszą ilość piany o dużej zawartości srebra
- W trakcie odsrebrzania do piany przechodzi cała zawartość Cu z ołowiu poddanego odsrebrzaniu. Czas odsrebrzania wynosi około 20h i limitowany jest przez szybkość grzania i chłodzenia ołowiu. Najbogatsza piana srebronośna jest z początkowej fazy ściągania. W związku z tym powinno się pierwsze 60-70% piany kierować do mufli likwacyjnej, natomiast z końcówki 30-40% piany powinno zawrócić do kotła odsrebrzającego w czasie napełniania. Takie postępowanie wzbogaca pianę w srebro i zmniejsza zużycie Zn. Bardzo istotnym czynnikiem jest temperatura rozpoczęcia procesu odsrebrzania, która powinna wynosić ok. 460-470 °C. W sposób widoczny utlenia Zn powiększając masę piany i stratę reagenta. Mieszadło w kotle powinno pracować na obrotach optymalnych.
- Odcynkowanie: W odsrebrzonym ołowiu pozostaje ok. 0,5% Zn. Usuwanie cynku prowadzi się w kotłach destylacyjnych. W wyniku destylacji próżniowej odparowane jest niemalże 100% cynku.
- Odbizmutowanie: Operacja odbizmutowania Pb przeprowadza się po zakończeniu usuwania Zn przy wynikowej zawartości Sb. Bizmut usuwa się z Pb za pomocą metalicznego wapnia i magnezu (proces Kroll-Bettertona). Można stosować stop MgCa zawierający 70% Mg i 30% Ca. Sb tworzy związki złożone z Mg, Ca i Bi przyczyniając się do zmniejszenia zużycia Ca i Mg. W procesie odbizmutowania Pb tworzą się trzy rodzaje związków: Ca2Bi2, CaMg2Bi2 i BiCa2Mg10Sb5. Wzajemny stosunek tych związków zależy od warunków przeprowadzenia procesu, Ca2Bi2 wydziela się w temperaturze 400-350 °C oraz dostatecznej ilości Ca i Bi, natomiast przy zawartościach Biponiżej 0,04% i w temp. powyżej 350 °C wydziela się podwójny bizmutek wapnia i magnezu. CaMg2Bi2. Związek BiCa2Mg10Sb5 tworzy się w niskich temperaturach i posiada bardzo małą rozpuszczalność w ołowiu.
- Odtalowanie: Odtalowanie przeprowadzane jest w tych samym kotle, co wcześniejszy proces odbizmutowania. Istotą rozwiązania jest zastosowanie do usuwania talu z ołowiu dodatku wapnia metalicznego w celu związania go w trudnotopliwe związki międzymetaliczne. W związku z silną tendencją talu do tworzenia kongruentnie topiących się związków międzymetalicznych z wapniem sprawdzono, że dodatek metalicznego wapnia do ciekłego ołowiupozwala na usuniecie z ołowiu talu. Przesłanką do takiego postępowania były diagramy równowag podwójnych Ca-Tl i Ca-Pb. Z powyższych diagramów wynika, że dodatek metalicznego wapnia do ciekłego ołowiu zanieczyszczonego talem będzie generował przebieg następujących reakcji chemicznych:
- reakcje z ołowiem:
Pb + n Ca = CanPb, gdzie n=1/3, 1, 2- reakcje z talem:
Tl + n Ca = CanTl, gdzie n=1/3, ¾, 1Temperatury topienia powyższych związków są znacznie wyższe od temperatury procesu. Przy założeniu, że temperatura procesu będzie wynosiła około 370 °C związki te utworzą stałą mieszaninę na powierzchni ciekłego ołowiu i będą mogły zostać łatwo usunięte. Metaliczny wapń może być do kąpieli dodawany albo w postaci granulowanej przy intensywnym mieszaniu, które zapobiegać będzie jego utlenianiu, albo w postaci gąsek, umieszczanych w odpowiednich koszach podwieszanych na mieszadle.- Usuwanie magnezu i wapnia: Celem rafinacji końcowej jest usunięcie z Pb: Ca i Mg oraz pozostałych z poprzednich procesów resztek: Zn, As, Sn, Sb. Dla pozbycia się tych zanieczyszczeń prowadzi się rafinację utleniającą:
-
- Ca + 2NaOH = CaO·Na2O + H2
- Mg + 2NaOH = MgO·Na2O + H2
Kolejność usuwania zanieczyszczeń jest zgodna z kolejnością podanych wyżej reakcji. Proces rozpoczyna się w temperaturze 450 °C, pamiętając, że ma charakter egzotermiczny.Odlewanie: Proces odlewania ołowiu prowadzi się w temp. 420-440 °C. Kocioł odlewniczy jest wyposażony w tubus z zaworem, który umożliwia całkowite spuszczenie z niego ołowiu i skierowanie na taśmę maszyny odlewniczej. Odlane gąski wymagają oczyszczenia ze zgarów. Celem szybkiego krzepnięcia odlanych gąsek stosuje się chłodzenie form wodą. Ołów rafinowany identyfikowany jest znakiem wytwórcy umieszczony w formie oraz numeratorem umieszczonym na końcu maszyny. - ZWIĄZKI OŁOWIU:




 -> Sole ołowiu:
-> Sole ołowiu:- a) Nieorganiczne sole ołowiu:
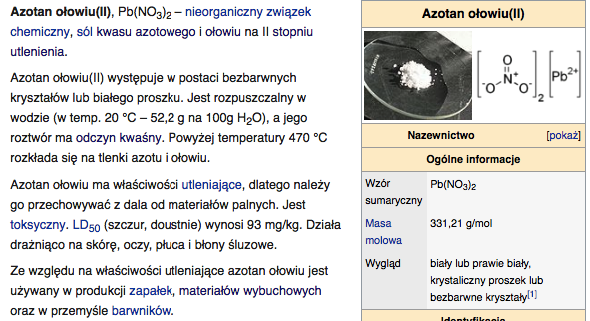











 b) Organiczne sole ołowiu:
b) Organiczne sole ołowiu:



 --> Związki ołowioorganiczne:
--> Związki ołowioorganiczne:


 6) Flerow (Fl, łac. Flerovium) - jest to pierwiastek chemiczny leżący w układzie okresowym w bloku p bezpośrednio pod ołowiem. Wszystkie znane izotopy są bardzo nietrwałe i nie występują naturalnie na Ziemi.
6) Flerow (Fl, łac. Flerovium) - jest to pierwiastek chemiczny leżący w układzie okresowym w bloku p bezpośrednio pod ołowiem. Wszystkie znane izotopy są bardzo nietrwałe i nie występują naturalnie na Ziemi.
Właściwości
Szczególnie interesujący jest nieodkryty dotąd (2009) izotop 298Fl ze 184 neutronami w jądrze. Model powłokowy budowy jądra atomowego przewiduje, że takie jądro będzie miało wyjątkowo długi czas życia.Właściwości chemiczne flerowu są jak dotąd słabo zbadane. Położenie w układzie okresowym wskazywałoby, że będzie on zachowywał się jak ciężki metal, przyjmując stopnie utlenienia II i IV. Tymczasem pierwsze wyniki badań sugerowały, że flerow niechętnie reaguje z innymi pierwiastkami, zachowując się raczej jak gaz szlachetny[6][9], ze względu na relatywistyczne przesunięcie energii stanów[9]. W 2012 roku przeprowadzono badania z wyższą czułością, których wyniki były sprzeczne z tymi rezultatami. Flerow jest lotnym pierwiastkiem, najmniej reaktywnym wśród węglowców, ale pomiary wskazywały, że jest bardziej reaktywny niż kopernik i jest metalem[10][11][12]. Sprzeczność wyników, ich mała liczba i duża niepewność pomiaru wciąż nie pozwalają na definitywne określenie charakteru metalicznego flerowu.



























Brak komentarzy:
Prześlij komentarz