
Azotowce (pniktogeny) - są to pierwiastki 15 (daw. VA lub V głównej) grupy układu okresowego. Są to azot, fosfor, arsen, antymon i bizmut. W warunkach normalnych, oprócz gazowego azotu, wszystkie występują w stanie stałym. Położenie w układzie okresowym wskazuje, że azotowcem jest także moscovium, wytworzone w ilości kilku atomów.


 1) Azot (N, łac. Nitrogenium) - jest to pierwiastek chemiczny o liczbie atomowej 7, niemetal z grupy 15 (azotowców) układu okresowego. Stabilnymi izotopami azotu są 14N i 15N. Azot w stanie wolnym występuje w postaci dwuatomowej cząsteczki N2. W cząsteczce tej dwa atomy tego pierwiastka są połączone ze sobą wiązaniem potrójnym. Azot jest podstawowym składnikiem powietrza(78,09% objętości), a jego zawartość w litosferze Ziemi wynosi 50 ppm. Wchodzi w skład wielu związków, takich jak: amoniak, kwas azotowy, azotynyoraz wielu ważnych związków organicznych(kwasy nukleinowe, białka, alkaloidy i wiele innych). Azot w fazie stałej występuje w sześciu odmianach alotropowychnazwanych od kolejnych liter greckich (α, β, γ, δ, ε, ζ). Najnowsze badania wykazują prawdopodobne istnienie kolejnych dwóch odmian (η, θ). Pierwiastek został odkryty w 1772 roku przez Daniela Rutherforda.
1) Azot (N, łac. Nitrogenium) - jest to pierwiastek chemiczny o liczbie atomowej 7, niemetal z grupy 15 (azotowców) układu okresowego. Stabilnymi izotopami azotu są 14N i 15N. Azot w stanie wolnym występuje w postaci dwuatomowej cząsteczki N2. W cząsteczce tej dwa atomy tego pierwiastka są połączone ze sobą wiązaniem potrójnym. Azot jest podstawowym składnikiem powietrza(78,09% objętości), a jego zawartość w litosferze Ziemi wynosi 50 ppm. Wchodzi w skład wielu związków, takich jak: amoniak, kwas azotowy, azotynyoraz wielu ważnych związków organicznych(kwasy nukleinowe, białka, alkaloidy i wiele innych). Azot w fazie stałej występuje w sześciu odmianach alotropowychnazwanych od kolejnych liter greckich (α, β, γ, δ, ε, ζ). Najnowsze badania wykazują prawdopodobne istnienie kolejnych dwóch odmian (η, θ). Pierwiastek został odkryty w 1772 roku przez Daniela Rutherforda.


(...)
Otrzymywanie azotu:
Azot o wysokiej czystości można uzyskać poprzez termiczny rozkład azotynu amonu, jednak taki azot zawiera małe ilości tlenków azotu:
- NH4NO2 → N2↑ + 2H2O

Najczystszy azot otrzymuje się przez rozkład termiczny w próżni azydku sodowego.
W laboratorium można otrzymać azot w wyniku łagodnego ogrzewania mieszaniny chlorku amonu (salmiaku) i azotynu sodu:
- NH4Cl + NaNO2 → N2↑ + NaCl + 2H2O

(...) W przemysłowej metodzie otrzymywania azotu skrapla się powietrze, stosując odpowiednie ciśnienie i temperaturę. W tych warunkach azot i wszystkie gazy znajdujące się nad nim na skali temperatur wrzenia skraplają się i są w otrzymanej cieczy. Trzy pierwiastki znajdujące się pod azotem: neon, wodór i hel pozostają w stanie gazowym i są przekazywane do oddzielnego procesu. Następny etap to frakcjonowanie poprzez podwyższenie temperatury (lub obniżenie ciśnienia), powodujące odparowanie ciekłego azotu. Odprowadzony azot jest powtórnie skraplany, przechowywany i transportowany w tzw. naczyniach Dewara lub w formie gazowej w temperaturze otoczenia w stalowych butlach. Jako pierwszy polską nazwę – azot – zaproponował Filip Walter. Używano też nazwy dusień i saletroród.




Wybrane związki azotu
- Nieorganiczne
- Tlenki azotu: tlenek diazotu (N2O), tlenek azotu (NO), tritlenek diazotu(N2O3), dwutlenek azotu (NO2), tetratlenek diazotu (N2O4), pentatlenek diazotu (N2O5)
- Związki z wodorem: amoniak, hydrazyna i azotowodór
- Kwasy: kwas azotowy i kwas azotawy
- Sole: azotany i azotyny, sole amonowe
- azotki
- Organiczne
- aminy
- amidy




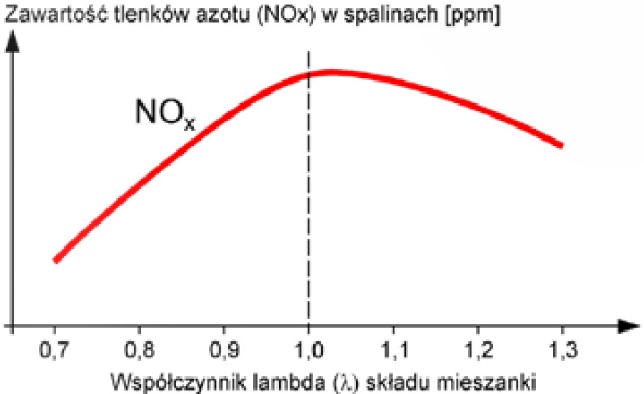



Źródło: intercars.com.pl; WKiŁ - Silniki poj. samochodowych.
Działanie tlenku azotu (II) na naczynia krwionośne:
 |
| Źródło: SynergyPoland.pl |

 |
| Źródło: sfd.pl |






(...)














-------------------------





































(...)
Właściwości azotu: Azot jest pierwiastkiem stosunkowo biernym chemicznie, co spowodowane jest bardzo wysoką energią wiązania potrójnego w cząsteczce N2, wynosi ona 945,33 ± 0,59 kJ·mol-1. Stopień dysocjacji w temperaturze 4000 K wynosi niecałe 3%. W podwyższonej temperaturze reaguje z metalamidając azotki, a także z innymi pierwiastkami, np. z wodorem tworzy amoniak, a z tlenem tlenki azotu, np.:
Procesy te (współcześnie zwłaszcza synteza amoniaku) wykorzystywane są przemysłowo do wiązania azotu atmosferycznego w celu produkcji licznych związków azotu. Innym wysokotemperaturowym procesem przemysłowym wiązania azotu jest reakcja azotu z karbidem prowadząca do cyjanamidu wapnia:
- CaC2 + N2 → CaCN2 + C
Naturalnie azot wiązany jest głównie przez bakterie azotowe w brodawkach roślin motylkowych oraz w trakcie wyładowań atmosferycznych.




 |
| Źródło: edupedia.pl - Obieg azotu w przyrodzie. |
Znaczenie biologiczne azotu: Należy do pierwiastków o bardzo dużym znaczeniu biologicznym. Wchodzi w skład wielu biocząsteczek, takich jak aminokwasy i białka, nukleotydy i kwasy nukleinowe. Większość organizmów nie jest zdolna do przyswajania azotu pierwiastkowego, z wyjątkiem bakterii azotowych wiążących wolny azot z powietrza, żyjących swobodnie w glebie lub symbiotycznych z roślinami bobowatymi (daw. motylkowatymi) bakterii brodawkowych. Dopiero w związkach, takich jak azotany, azotyny lub sole amonowe, jest przyswajalny przez rośliny.
Wpływ azotu na organizm:
-> Azot atmosferyczny: Azot pod normalnym ciśnieniem jest obojętny dla organizmów żywych. Może jednak wywołać objawy zatrucia u osób przebywających w powietrzu o zwiększonym ciśnieniu. W takich warunkach azot lepiej rozpuszcza się w płynach ustrojowych i tkankach bogatych w lipidy (np. w mózgu), co prowadzi do pojawienia się objawów zatrucia, takich jak:
- przy ciśnieniu 4 atm
- euforia, skłonność do śmiechu, gadulstwo
- spowolnienie reakcji na bodźce
- przy ciśnieniu 10 atm
- ostre zaburzenia pracy mięśni, koordynacji ruchów
- zawroty głowy
- zaburzenia świadomości
- przy ciśnieniu powyżej 10 atm
- po kilku minutach następuje utrata świadomości i śpiączka
Jeszcze bardziej niebezpieczne od rozpuszczania się azotu w tkankach jest jego wydzielanie się w postaci pęcherzyków gazu podczas zmniejszania ciśnienia. Prowadzić to może do groźnej dla zdrowia i życia choroby dekompresyjnej (kesonowej).

-> Związki azotu w pożywieniu i wodzie pitnej: Woda zawierająca więcej niż 45 ppm związków azotu jest uznawana za szkodliwą dla dzieci. Szczególnie toksyczne są azotyny. Ich obecność w glebie i wodzie pitnej jest jednak znikoma. Dość niebezpieczne dla środowiska jest składowanie związku azotu np. nawozów w zbiornikach z blachy ocynkowanej ze strony wewnętrznej, gdyż związki azotu wchodzą w reakcje z związkami cynku i żelaza, a po zastosowaniu takiego nawozu powstałe związki przedostają się do wód gruntowych.
-> Azot w glebie: Azot wolny z atmosfery nie jest przyjmowany przez rośliny, ponieważ wymagają one związków, dodatkowo 99% azotu glebowego pozostaje w związkach organicznych o zbyt złożonej budowie, by mogła je przyjąć większość gatunków roślin. Azotu, który może zostać przyjęty przez roślinny wyższe, jest przeciętnie mniej niż 34 kg/ha, a azotu w związkach – około 3385 kg/ha. Ilość azotu przyjmowanego może wynieść nawet do 112,5 kg/ha.
-> Wpływ azotu na wzrost roślin: Azot jest potrzebny roślinom głównie w fazie wzrostu, ze względu na możliwość akumulacji azotu przez roślinę, przy wysokim stężeniu azotu w glebie, absorpcja tego pierwiastka jest znacznie szybsza niż wzrost rośliny. Przy niedoborze azotu rośliny rosną wolno, są słabe, bledsze. Kolor ten jest związany z deficytem chlorofilu, który bierze udział w procesie fotosyntezy. Skrajny niedobór azotu może powodować żółtawobrązowe zabarwienie fragmentów liści. Niedobór azotu w glebie może zostać uzupełniony przez nawożenie nawozami azotowymi lub poprzez zmianę uprawy na potrzebującą mniej azotu. Nadmiar azotu nie jest w zasadzie szkodliwy dla rośliny, gdyż rośliny mogą sobie go akumulować, jednakże nadmiar związków azotu może powodować nadmierny wzrost rośliny i brak możliwości przyjmowania innych pierwiastków niezbędnych roślinie. Groźny jest za to nadmiar azotu połączony z niedoborem fosforu, potasu lub wody. Nadmiar azotu może szkodzić jakości i wielkości plonów. Nadmiar azotu jest szkodliwy dla drzew w przypadku mrozów. Rośliny mające za dużo azotu są ciemnozielone, wyglądają aż nazbyt dorodnie. Gleba posiadająca za dużo azotu powinna być nawożona nawozami zawierającymi potas i fosfor.
-> Zastosowanie azotu: Ciekły azot jest stosowany jako środek chłodzący do uzyskiwania temperatur poniżej -100 °C. W postaci gazowej azot wykorzystywany jest jako najtańsza z dostępnych atmosfer ochronnych w wielu procesach przemysłowych, a także jako gaz roboczy w niektórych układach pneumatycznych. Z azotu otrzymuje się amoniak oraz tlenki azotu wykorzystywane w produkcji kwasu azotowego, związki o dużym znaczeniu przemysłowym. Ponadto szeroko wykorzystuje się azotany, azotyny, hydrazynę, hydroksyloaminę i in. związki zawierające azot.











ZWIĄZKI AZOTU:




















































*Związki azotu omówić w oddzielnym poście.
 2) Fosfor (P, gr. phosphoros - "niosący światło", łac. phosphorus) - jest to pierwiastek chemiczny, niemetal. Jedynym stabilnym izotopem fosforu jest 31P. Fosfor występuje w czterech odmianach alotropowych, jako: fosfor biały, czerwony, fioletowy oraz czarny.
2) Fosfor (P, gr. phosphoros - "niosący światło", łac. phosphorus) - jest to pierwiastek chemiczny, niemetal. Jedynym stabilnym izotopem fosforu jest 31P. Fosfor występuje w czterech odmianach alotropowych, jako: fosfor biały, czerwony, fioletowy oraz czarny.
Fosfor biały: W wodzie jest prawie nierozpuszczalny, słabo rozpuszcza się w etanolu i chloroformie, dobrze w dwusiarczku węgla. Z ostatniego z wymienionych rozpuszczalników wydziela się po odparowaniu w postaci kryształów układu regularnego o połysku diamentowym. Przy zwykłym ciśnieniu fosfor biały topi się w 44 °C, a wrze w 277 °C przechodząc w pary o składzie P4. Ponieważ bardzo łatwo ulega samozapłonowi, przechowuje się go zawsze pod wodą. Jest trudny do ugaszenia, ponieważ w wysokich temperaturach reaguje z wodą, która go dodatkowo rozprasza. Ma tendencję do przechodzenia w inne, bardziej trwałe odmiany, do których należy fosfor czerwony. Cząsteczka fosforu białego składa się z 4 atomów. W temperaturze pokojowej ulega powolnemu utlenianiu, emitując światło (chemiluminescencja)[9]. Był stosowany jako składnik farb, które nanoszono na wskazówki zegarków (świeciły światłem widocznym w ciemności).
Reaguje z wrzącym roztworem wodorotlenku barowego:
2P4 + 3Ba(OH)2 → 2PH3↑ + 3Ba(H2PO2)2
W reakcji powstaje fosforiak (fosforowodór) i barowa sól kwasu fosforowego(I).





Jest skrajnie łatwopalny. Z tego powodu dawniej produkowano z niego zapałki. Były to jednak zapałki bardzo niebezpieczne, ponieważ zapalały się po potarciu o każdą szorstką powierzchnię, a ponadto były trujące. Obecnie jedynie szorstka powierzchnia pudełek, służących do pocierania zapałek, zawiera nieco czerwonego fosforu, zmieszanego z tłuczonym szkłem i dwutlenkiem manganu. Wskutek ogrzania, wywołanego potarciem zapałki o tę powierzchnię, fosfor się zapala i płomień przenosi się na główkę zapałki. Jest silnie trujący; dla człowieka o wadze 60 kg śmiertelna dawka wynosi 100 mg. Fosfor biały stosowany jest jako broń zapalająca i trutka na szczury.





(...)
Fosfor czerwony: Inaczej zwany fosforem bezpostaciowym. Otrzymuje się go przez prażenie fosforu białego w atmosferze azotu i temperaturze 250 °C z użyciem jodujako katalizatora. Jest nietoksyczny, mniej reaktywny od fosforu białego. Zapala się powyżej 400 °C, jego ugaszenie również jest bardzo trudne, ponieważ pali się nawet przy ograniczonym dostępie tlenu. Jest używany jako jeden ze składników draski na pudełkach od zapałek.

Fosfor fioletowy: Powstaje w wyniku ogrzewania fosforu czerwonego w próżni w temperaturze ok. 550 °C. Inna metoda otrzymywania tej odmiany to krystalizacja z roztworu fosforu białego w stopionym ołowiu. Nierozpuszczalny w żadnej substancji. Mało aktywny chemicznie.









(...)
Fosfor czarny: Najtrwalsza odmiana fosforu. Otrzymywany przez ogrzewanie fosforu białego bez dostępu tlenu w temp. 220 °C i pod ciśnieniem 12 tys. atm. Ma on barwę szarą, połysk metaliczny, przewodzi prąd elektryczny.




Istnieją sprzeczne informacje co do tzw. luksusowej konsumpcji, czyli pobierania przez rośliny fosforu w ilościach większych niż wymagane. Według danych głównie opartych na obserwacji roślin uprawnych podawane jest, że zjawisko to w zasadzie nie zachodzi, choć rośliny żyjące na glebach żyznych mogą zawierać w tkankach więcej fosforu niż rośliny siedlisk ubogich, z kolei według obserwacji roślin mokradłowych fosfor należy do biogenów pobieranych w sposób luksusowy, czemu sprzyjają mechanizmy magazynowania go w postaci związków fosfoorganicznych. U tych gatunków roślin, które nie potrafią aktywnie wydalać nadmiaru fosforu, może dochodzić do zatrucia jego nadmiarem. Związki fosforu są dość ruchliwe wewnątrz roślin i są przemieszczane między organami w zależności od zapotrzebowania. W tkankach roślinnych występuje zarówno w postaci nieorganicznej (głównie jako fosforany, mniej jako pirofosforany), jak i organicznej. Organiczne związki fosforu to m.in. kwasy zawierające grupy karboksylowe i fosforanowe czy estry. Kwas 3-fosfoglicerynowy i aldehyd 3-fosfoglicerynowy to estry biorące udział w oddychaniu i fotosyntezie. Fosfolipidy, w tym lecytyny, budują błony komórkowe, a fityna ma charakter substancji zapasowej, magazynującej fosfor. Substancje te z kolei przyswajane z pokarmem przez zwierzęta pełnią w ich organizmach dodatkowe funkcje. Grupa fosforanowa wchodzi także w skład nukleotydów biorących udział w energetyce komórki (np. ATP), komunikacji komórek (np. cAMP) oraz w przechowywaniu informacji genetycznej i syntezie białek (DNA i RNA).
Fosfor obok azotu i potasu jest jednym z trzech głównych makroelementów potrzebnych do życia roślin (poza tzw. organogenami, czyli węglem, wodorem i tlenem). Niedobór fosforu dostępnego dla roślin powoduje u nich różnego rodzaju objawy niedożywienia – spowolnienie wzrostu, rachityczność, mniejszą ilość syntetyzowanych substancji (np. witamin). Stąd też wiele nawozów sztucznych to nawozy fosforowe lub nawozy NPK. Jednocześnie silne nawożenie związkami azotu bez suplementacji fosforowej może zmniejszać przyswajalność glebowego fosforu dla roślin. W przypadku roślin wieloletnich nawożenie na glebach ubogich powoduje przez kilka pierwszych lat zwiększony pobór i koncentrację w tkankach, po czym następuje wysycenie.
Fosfor jest jednym z głównych makroelementów w organizmach, ale w wodach jest mikroelementem. Ponieważ zawartość fosforu w siedlisku jest stosunkowo mała (wielokrotnie niższa niż w roślinach), często jest ona czynnikiem limitującym wzrost roślin i innych organizmów samożywnych w danym środowisku, zgodnie z prawem minimum Liebiga. Dotyczy to zwłaszcza wód słodkich. Dopływ związków fosforu i innych biogenów do wód powoduje ich eutrofizację. Zwiększenie ilości fosforu w jeziorach skutkuje zwiększoną produkcją pierwotną, jak również przebudową składu gatunkowego zespołów glonów i makrofitów (często zwiększeniem udziały sinic). Ponieważ fosfor trafia do obiegu w organizmach, jego wykrywalna zawartość w wodzie może nadal być stosunkowo niska. W środowiskach lądowych w pewnych warunkach fosfor również może być czynnikiem limitującym produkcję pierwotną. Powolna eutrofizacja to proces naturalny, ale jest znacznie przyspieszana przez zanieczyszczenia antropogeniczne, głównie ścieki komunalne z detergentami zawierającymi fosforany i spływy z nawożonych pól.
Zastosowanie: Tlenki fosforu używane są jako reduktory (P4O6) lub środki suszące (P4O10). Kwas fosforowy (H3PO4) jest dodatkiem do napojów gazowanych typu cola. Związki tego pierwiastka wykorzystywane są również w przemyśle chemicznym jako katalizatory. Fosforan sodu stosuje się do proszków do prania; okazało się, że coraz powszechniejsze używanie detergentów powoduje zwiększanie stężenia związków fosforu w wodach rzek i jezior, zwłaszcza w Polsce, gdzie nie wszystkie ścieki są oczyszczane. Wynikiem tego jest eutrofizacja wód. Fosfor jest również składnikiem lutu twardego (np. L-CuP6) stosowanego do lutowania rur miedzianych w instalacjach wodnych, gazowych lub freonowych.

Występowanie w przyrodzie: Ważne minerały fosforu to apatyt i fosforyty. Fosfor tworzy szereg kwasów, z czego najważniejszym jest kwas fosforowy H3PO4. Związki fosforu są stosowane jako nawozy sztuczne. Postać związków fosforu w wodach zależy od jej odczynu. W wodach o pH<6 przeważają jony H2PO4−, a w wodach o pH>7 HPO42−. Jony PO43− występują w małych ilościach jedynie w wodach zasadowych (pH>8,5), a kwas H3PO4 w wodach kwaśnych (pH<6).
Zawartość fosforu w wodach gruntowych wynosi od 24,10 μg/l w strefie tropikalnej do 80,00 μg/l na obszarach górskich (średnio w strefie ługowania skał 61,00 μg/l). Fosfor w wodach podziemnych pochodzi on głównie z wietrzenia apatytów, a także z zanieczyszczeń kopalnianych. Ze względu na niewielką ruchliwość związków fosforu, w wodach podziemnych stosunkowo mało jest fosforu pochodzącego z zanieczyszczeń komunalnych, inaczej niż w wodach powierzchniowych. W glebie fosfor w większości występuje nie w formie rozpuszczonej, lecz stałej. W warunkach środkowoeuropejskich wśród gleb najbogatsze w fosfor są czarnoziemy, a najuboższe bielice. Jeszcze uboższe są natomiast piaski. Fosfor w glebach silnie kwaśnych (pH<5,5) występuje głównie w postaci fosforanów żelaza, glinu i manganu, a w pozostałych – fosforanów wapnia i magnezu (hydroksyapatyt, fluoroapatyt). Od kilku do ¾ fosforu glebowego to fosfor w związkach organicznych (głównie fityna i fityniany) pochodzący z rozkładu szczątków organizmów.
Historia: Fosfor został odkryty przez niemieckiego alchemika Henniga Branda w Hamburgu w 1669 r. podczas długotrwałego prażenia moczu. W pierwszej komercyjnej metodzie otrzymywania fosforu (koniecznego do produkcji zapałek) substancją wyjściową były kości zwierzęce, co znalazło odbicie w proponowanej polskiej nazwie fosforu koścień.



Kwasy zawierające fosfor
Znanych jest wiele kwasów tlenowych zawierających fosfor (w niektórych przypadkach znane tylko jako sole lub estry), np.:
- kwas fosfinowy (fosforowy(I), podfosforawy) – H3PO2
- kwas fosfonowy (fosforawy, fosforowy(III)) – H3PO3
- kwas metafosforawy (metafosforowy(III)) – HPO2
- kwas metatrifosforawy (metatrifosforowy(III)) – H3P3O6 (związek cykliczny)
- kwas difosfonowy (pirofosforawy, difosforowy(III)) – H4P2O5
- kwas podfosforowy (fosforowy(IV)) – H4P2O6
- kwas fosforowy (fosforowy(V), ortofosforowy) – H3PO4 (najczęściej spotykany kwas fosforu)
- kwas difosforowy (pirofosforowy, difosforowy(V)) – H4P2O7
- kwas trifosforowy (ortotrifosforowy(V)) – H5P3O10
- kwas metafosforowy – wzór empiryczny: HPO3
- kwas metatrifosforowy – H3P3O9
- kwas metatetrafosforowy – H4P4O12
- kwas nadtlenofosforowy
Znane są też liczne analogi powyższych kwasów, w których jeden lub więcej atomów tlenu zostało zastąpionych przez atomy innych tlenowców(S, Se, Te), azotu lub boru[21], np.:
- kwas tiofosfonowy H3PO2S
- kwas selenofosforowy H3PO3Se
- kwas tellurofosforowy H3PO3Te
- kwas amidofosforowy H3PO3NH2
- kwas boranofosforowy H3PO3BH3
Ponadto otrzymano szereg mieszanych polikwasów zawierających jako atomy centralne fosfor i metale przejściowe, np. kwasy molibdenowanadofosforowe H4[PMo11VO40]•34H2O, H5[PMo10V2O40]•32H2O i H6[PMo9V3O40]•34H2O występujące w postaci pomarańczowych kryształów[22].
W kwasach fosforu zawierających wiązanie P-H (np. w kwasie fosfonowym) atomy wodoru mogą zostać zastąpione resztami organicznymi, tworząc liczną grupę kwasów alkilo- i arylo-fosfonowych i fosfinowych oraz ich analogów zawierających inne heteroatomy[21], np. kwas dodecylofosfonowy.
Częściowo zestryfikowane kwasy fosforu zawierające labilne atomy wodoru, mają także charakter kwasowy, np. kwas rybonukleinowy (RNA) i kwas deoksyrybonukleinowy (DNA).



Fosforanowanie (fosfatyzacja, daw. parkeryzacja) – proces chemicznegolub elektrochemicznego wytwarzania ochronnej matowo-szarej powłoki fosforanów na powierzchni metali (gł. stali); prowadzony w gorących roztworach fosforanów i kwasu fosforowego. Powłoka fosforanowa jest skuteczną powłoka antykorozyjną, odporną na działanie wysokich temperatur, bardzo dobrze niweluje refleksy świetlne, jednak jest stosunkowo miękka. Poza tym zmniejsza współczynnik tarcia oraz stanowi dobry podkład dla farb i lakierów. Występuje również w barwie ciemnoszarej i czarnej. Może być stosowana zarówno w nożach ze stali narzędziowej, jak i nierdzewnej.


ZWIĄZKI FOSFORU:

-> Kwasy fosforu:

--> Tlenowe kwasy fosforu:







---> Kwasy fosfonowe:





















--> Nieorganiczne związki fosforu:














 3) Arsen (As, łac. Arsenium) - jest to pierwiastek chemiczny z grupy azotowców o właściwościach metaloidu. Występuje w skorupie ziemskiej w ilości 2,5 ppm(zajmując 20. miejsce pod względem rozpowszechnienia wśród pierwiastków). Tworzy ponad 200 minerałów, z których najbardziej rozpowszechnione są: arsenopiryt, lelingit, aurypigment, realgar. Często towarzyszący także złożom siarczkowym innych metali, np. pirytom, chalkopirytom, galenie, blendzie cynkowej. W naturze arsen występuje również w stosunkowo dużej liczbie związków arsenoorganicznych. Są to np. kwas kakodylowy i inne kwasy arsenoorganiczne, organiczne pochodne arsyny, arsenowe analogi biocząsteczek, np. arsenobetaina lub arsenocholina.
3) Arsen (As, łac. Arsenium) - jest to pierwiastek chemiczny z grupy azotowców o właściwościach metaloidu. Występuje w skorupie ziemskiej w ilości 2,5 ppm(zajmując 20. miejsce pod względem rozpowszechnienia wśród pierwiastków). Tworzy ponad 200 minerałów, z których najbardziej rozpowszechnione są: arsenopiryt, lelingit, aurypigment, realgar. Często towarzyszący także złożom siarczkowym innych metali, np. pirytom, chalkopirytom, galenie, blendzie cynkowej. W naturze arsen występuje również w stosunkowo dużej liczbie związków arsenoorganicznych. Są to np. kwas kakodylowy i inne kwasy arsenoorganiczne, organiczne pochodne arsyny, arsenowe analogi biocząsteczek, np. arsenobetaina lub arsenocholina.







Arsen ma dwie odmiany alotropowe:
- α – jest kruchym metalem, który matowieje w kontakcie z powietrzem i silnie reaguje z wodą;
- β – jest żółtym, bezpostaciowym proszkiem, dużo mniej reaktywnym od odmiany α.
Zastosowanie arsenu: Związki arsenu były znane od starożytności. W formie czystej wyodrębnił go jako pierwszy (prawdopodobnie) alchemik Albert Wielki w 1250, choć do tego odkrycia pretendują też starsi alchemicy arabscy i chińscy lekarze ludowi.

*Św. Albert Wielki, Albert z Kolonii, Albert z Lauingen, łac. Albertus Magnus(ur. 1193/1205? w Lauingen (Donau), zm. 15 listopada 1280 w Kolonii) – teolog, dominikanin, filozof scholastyczny. Znany także z obszernej wiedzy przyrodniczej, przypisuje mu się także zajmowanie się alchemią. Święty Kościoła katolickiego, doktor Kościoła (znany jako doctor universalis lub doctor expertus).
Albert jako jeden z pierwszych w systematyczny sposób łączył filozofię Arystotelesa z teologią katolicką – w Arystotelesie dostrzegł możliwość pogodzenia wiedzy przyrodniczej i wiary. Jako jeden z pierwszych głosił też odrębność metody teologii od metody nauk przyrodniczych oraz inny charakter prawd wiary i prawd naukowych. Te podstawy metodologiczne umożliwiły mu znaczne poszerzenie wiedzy w dziedzinie botaniki. Wszystkie te założenia przejął jego największy uczeń, Tomasz z Akwinu.
Albert pozostaje pod wpływem Awicenny, któremu zawdzięcza zbliżoną do emanacyjnej koncepcję stworzenia i pojęcie Boga jako pierwszej przyczynystwarzającej hierarchię bytów, a także – przy całym swoim głębokim arystotelizmie – pewne skłonności neoplatońskie.
Św. Albert żywo interesował się każdą dostępną w jego czasach dziedziną wiedzy przyrodniczej, dzięki czemu stał się (obok Rogera Bacona) najbardziej uczonym człowiekiem swoich czasów – ze względu na tę rozległość zainteresowań potomni nadali mu tytuł Doctor Universalis. Mimo tego część tej wiedzy jest w typowy dla jego czasów sposób zepsuta – wydaje się nawet, że uważał Platona i Speuzypa za stoików.
Większość swojej wiedzy w zakresie sztuk wyzwolonych zaczerpnął z naukowych pism Arystotelesa, stanowiących fundament wiedzy przyrodniczej jego czasów. Nie oznacza to jednak – wbrew potocznym sądom żywionym nieraz o Średniowieczu – że jest bezkrytyczny. Mówi np., że „Celem nauk przyrodniczych nie jest proste przyjmowanie wyników [narrata] uzyskanych przez innych, ale poszukiwanie przyczyn które działają w naturze” (De Miner., lib. II, tr. ii, i). W swoim traktacie o roślinach głosi zasadę, że experimentum solum certificat in talibus [w badaniach jedynym bezpiecznym przewodnikiem jest doświadczenie]
Szczególnie interesował się botaniką i chemią. W 1250 otrzymał arszenik. Współcześni oskarżali go o uprawianie magii, częściowo na skutek zazdrości, częściowo na skutek niezrozumienia jego nieraz bardzo nowatorskich idei (stworzył np. androida, maszynę o postaci człowieka). Przypisywane mu często grymuary mają jednak – mimo czarnej legendy jaka je otacza – wyjątkowo niewinny charakter. W swym dziele Compositium de compositis opisał m.in. że związek otrzymany z rozpuszczenia srebra w kwasie azotowym „... barwi ręce ludzkie na czarno, ciężką do pozbycia się farbą...” zauważając tym samym zjawisko światłoczułości związku srebra, co zbadane zostało dokładniej dopiero kilkaset lat później...
(...) W czasach starożytnych mieszaniny zawierające aurypigment i realgar stosowano do leczenia chorób płuc i skóry. W późniejszych wiekach znaczenie arsenu w medycynie zaczęło znacznie rosnąć. W XVIII i XIX w. arsen stał się wręcz podstawą ówczesnej farmakologii. Preparaty arsenowe, stosowane w przeróżnej postaci od past, roztworów, tabletek począwszy, na zastrzykach dożylnych i podskórnych skończywszy, używano do zwalczania większości chorób: reumatyzmu ,astmy, malarii, gruźlicy, cukrzycy, śpiączki afrykańskiej, nadciśnienia, wrzodów żołądka, zgagi, egzemy, łuszczycy, a nawet białaczki.

Na przełomie XIX i XX w. zaczęto stosować związki arsenoorganiczne, które okazały się być znacznie mniej toksyczne dla ludzi i zwierząt niż związki nieorganiczne. W drugiej połowie ubiegłego wieku wycofano jednak z obiegu, mimo ich dużej skuteczności, niemal wszystkie leki arsenowe (głównie za sprawą właściwości rakotwórczych). Obecnie poza melarsoprolem, acetarsolem, salwarsolem i trójtlenkiem arsenu nie stosuje się w lecznictwie związków arsenu. W ostatnich latach zaczyna wzrastać zainteresowanie arszenikiem, jako środkiem w terapii przeciwnowotworowej.

---------------
(...) Poza medycyną arsen znalazł zastosowanie w produkcji półprzewodników (jako arsenek galu), polepszania jakości niektórych stopów (m.in. 0,5-2% dodatek do ołowiu poprawia sferyczność amunicji ołowianej, a 0,1-3% dodatek do stopów łożyskowych zawierających ołów zwiększa ich wytrzymałość), w produkcji bojowych środków trujących (luizyty), przy impregnacji drewna i jako dodatek do szkła (dając mu zielonkawą poświatę).









 (...) Przez wiele lat związki arsenu stosowane były również w garbarstwie oraz jako pigmenty i środki ochrony roślin(głównie arseniany), jednak ich znaczenie w ostatnich latach zmalało znacznie na rzecz mniej toksycznych odczynników.
(...) Przez wiele lat związki arsenu stosowane były również w garbarstwie oraz jako pigmenty i środki ochrony roślin(głównie arseniany), jednak ich znaczenie w ostatnich latach zmalało znacznie na rzecz mniej toksycznych odczynników.
*Arseniany (nazwa systematyczna: tetraoksydoarseniany(3−); w systemie Stocka: arseniany(V)) – sole i estry kwasu arsenowego.
 Przed garbowaniem usuwano resztki zasad za pomocą kwasowych kąpieli z otrąb (fermentacja) oraz wytrawiano nawozem (kurzym lub gołębim). Zawarte w nim enzymy proteolityczne hydrolizowały białko skórne, zmydlały tłuszcz, usuwały wapno. Skóra po zabiegu była bardziej ciągliwa i miękka. W czasach wcześniejszych w celu usunięcia włosia stosowano tzw. pocenie skóry. Jest to metoda polegająca na celowym dopuszczeniu do zapoczątkowania procesów gnilnych, przy których następuje rozluźnienie nasady włosa i jego wychodzenie, podczas gdy włókna tkanki skórnej pozostawały jeszcze nienaruszone. Metoda ta stosowana jest do dzisiaj przy skórach zwartych (siodlarstwo, futrzarstwo). Skóra ta przy wysokiej wodoodporności jest bardzo wytrzymała na rozciąganie. Można ją zabezpieczyć metodami zarówno fizycznymi, jak i chemicznymi.
Przed garbowaniem usuwano resztki zasad za pomocą kwasowych kąpieli z otrąb (fermentacja) oraz wytrawiano nawozem (kurzym lub gołębim). Zawarte w nim enzymy proteolityczne hydrolizowały białko skórne, zmydlały tłuszcz, usuwały wapno. Skóra po zabiegu była bardziej ciągliwa i miękka. W czasach wcześniejszych w celu usunięcia włosia stosowano tzw. pocenie skóry. Jest to metoda polegająca na celowym dopuszczeniu do zapoczątkowania procesów gnilnych, przy których następuje rozluźnienie nasady włosa i jego wychodzenie, podczas gdy włókna tkanki skórnej pozostawały jeszcze nienaruszone. Metoda ta stosowana jest do dzisiaj przy skórach zwartych (siodlarstwo, futrzarstwo). Skóra ta przy wysokiej wodoodporności jest bardzo wytrzymała na rozciąganie. Można ją zabezpieczyć metodami zarówno fizycznymi, jak i chemicznymi.

Na przełomie XIX i XX w. zaczęto stosować związki arsenoorganiczne, które okazały się być znacznie mniej toksyczne dla ludzi i zwierząt niż związki nieorganiczne. W drugiej połowie ubiegłego wieku wycofano jednak z obiegu, mimo ich dużej skuteczności, niemal wszystkie leki arsenowe (głównie za sprawą właściwości rakotwórczych). Obecnie poza melarsoprolem, acetarsolem, salwarsolem i trójtlenkiem arsenu nie stosuje się w lecznictwie związków arsenu. W ostatnich latach zaczyna wzrastać zainteresowanie arszenikiem, jako środkiem w terapii przeciwnowotworowej.

Historia
W XV wieku William Withering stwierdził, że trójtlenek arsenu stosowany w małych dawkach wykazuje działanie lecznicze[5]. W XVIII wieku Thomas Fowler sporządził 1% roztwór arszeniku i węglanu potasu, który stosowany był w leczeniu chorób skóry (przede wszystkim łuszczycy) aż do XX wieku[7]. Powstał również lek oparty na bazie arsenu – salwarsan – stosowany w leczeniu kiły, zsyntetyzowany przez Paula Ehrlicha. Został on jednak wyparty przez penicylinę[8]. Związki arsenu były powszechnie wykorzystywane w leczeniu różnych chorób w XIX i na początku XX wieku[7][9][10].
Pierwsze doniesienia o aktywności przeciwnowotworowej trójtlenku arsenu pochodzą z 1878 roku, kiedy to w raporcie Boston City Hospital opisano działanie roztworu Fowlera obniżające poziom leukocytów we krwi u dwóch zdrowych osób i jednego chorego[5][11]. Trójtlenek arsenu był stosowany w leczeniu białaczki aż do momentu wprowadzenia radioterapii. Powrócił do łask w latach 30. XX wieku, kiedy to pojawiły się pierwsze badania potwierdzające wysoką skuteczność trójtlenku arsenu w leczeniu przewlekłej białaczki szpikowej[12].
Pod koniec lat 60. XX w. lekarzy pracujących w Akademii Medycznej w Harbin w Chinach wysłano do ośrodka zajmującego się tradycyjną medycyną chińską. Stosowano tam maść na czerniaka, której głównym składnikiem był arszenik. W tamtych czasach arsenał leków przeciwnowotworowych był bardzo ograniczony, wobec czego lekarze zaczęli stosować arszenik. Pierwsze próby opierały się na doustnym podawaniu tej substancji, wykazywała ona jednak silne działanie toksyczne. W marcu 1971 rozpoczęto pierwsze próby, w których arszenik podawano dożylnie. W takiej postaci wykazywał znacznie mniejszą toksyczność. Przez wiele lat tlenek arsenu(III) podawano chorym z różnymi nowotworami, najlepsze efekty uzyskując w leczeniu ostrej białaczki promielocytowej[13]. Spośród pacjentów uczestniczących w pierwszej serii prób w Harbin więcej niż połowa przeżyła 5 lat. Dzięki temu badania rozszerzono na inne ośrodki w Chinach[14][15], a następnie zorganizowano je w Sloan-Kettering Memorial Institute w Nowym Jorku[16]. Wyniki badań klinicznych okazały się na tyle korzystne, że w 2000 roku lek uzyskał aprobatę FDA[17].
Mechanizm działania[edytuj | edytuj kod]







Mechanizm działania trójtlenku arsenu jest złożony i nie do końca poznany. Ogólnie rzecz ujmując, lek hamuje proliferację komórek nowotworowych oraz doprowadza do ich różnicowania i/lub apoptozy, która przebiegać może w różny sposób, w zależności od zaangażowanych w nią organelli komórkowych i procesów biochemicznych. Trójtlenek arsenu indukuje apoptozę na drodze:
- oddziaływania z receptorami błony komórkowej (ścieżka zewnętrzna)
- oddziaływania z mitochondriami (ścieżka wewnętrzna)
Pierwsza z tych dróg polega na połączeniu ligandu z receptorem znajdującym się na powierzchni błony komórkowej. Zespolenie tych dwóch cząstek prowadzi do aktywacji wielu różnych genów oraz uwalnia kaskadę białek, charakterystycznych dla procesu apoptozy[25].
Trójtlenek arsenu oddziałuje również na mitochondria. Jedną z pierwszych zmian w ich strukturze, wywołaną działaniem leku, jest otwarcie megakanałów i wypływ tzw. białek śmierci, przede wszystkim cytochromu C, APAF-1 (czynnik aktywacji proteaz apoptotycznych, ang. apoptotic peptidase activating factor 1), czynnika AIF, białka Smac/DIABLO i endonukleazy z przestrzeni międzybłonowej mitochondriów do cytozolu. W cytoplazmie powstaje kompleks białek zwany apoptosomem, który uruchamia dalsze procesy prowadzące do apoptozy[26].
Bez względu na to, czy apoptoza indukowana jest zewnętrznie czy wewnętrznie, zawsze biorą w niej udział kaspazy, których aktywacja nieodwracalnie prowadzi komórkę na drogę programowanej śmierci[27][28]. Ponadto apoptoza regulowana jest przez białka z rodziny Bcl-2, które działać mogą zarówno pro-, jak i antyapoptotycznie[29].
Przyczyną występowania ostrej białaczki promielocytarnej jest translokacja genu kodującego receptor kwasu retinowego (RAR α) z chromosomu 17 w pobliże genu PML, znajdującego się na chromosomie 15. Prowadzi to do fuzji genów i powstania produktu PML/RAR α[30]. Białko to hamuje różnicowanie oraz śmierć komórek, w których się znajduje. Trójtlenek arsenu, już w małych stężeniach, powoduje rozpad PML/RAR α, przywracając w ten sposób (częściowo) różnicowanie zmienionych nowotworowo promielocytów[31]. Trójtlenek arsenu aktywuje kinazę JNK, zwaną również kinazą N-końca białka c-jun (ang. c-jun N-terminal kinase) lub kinazą aktywowaną stresem (ang. stress-activated protein kinase– SAPK), należącą do rodziny kinaz MAPK (ang. mitogen-activated protein kinases). Enzymy te mają kluczowe znaczenie dla przekazywania sygnałów w komórce. W normalnych warunkach kinaza JNK aktywowana jest przez fosforylację reszt treoniny i tyrozyny[32][33]. W badaniach na określonych liniach komórkowych pobranych od chorych z ostrą białaczką promielocytową udowodniono jednak, że wspomniana aktywacja zachodzi również pod wpływem trójtlenku arsenu[34].
Wydaje się, że aktywacja kinazy JNK prowadzi do fosforylacji zarówno białek antyapoptotycznych (Bcl-2, Bcl-Xl), jak i białek proapoptotycznych – Bax (ang. Bcl-2-associated X protein), Bak (ang.Bcl-2 homologous killer) i Bid (ang. BH3 interacting domain death agonist), czyli de facto do ich uczynnienia. Białka proapoptotyczne zawierają domenę BH3, która odpowiada za ich "śmiercionośną" aktywność. Powodują one powstawanie kanałów jonowych w błonie mitochondrium, czego wynikiem jest wypływ wspomnianych już czynników apoptotycznych do cytoplazmy. Białka antyapoptotyczne zawdzięczają swoje działanie hydrofobowej szczelinie w ich strukturze przestrzennej, która wiąże się z domeną BH3, neutralizując tym samym działanie "śmiercionośnych" białek. W normalnych warunkach o apoptozie komórki decyduje stosunek ilości białek proapoptotycznych do antyapoptotycznych. W przypadku apoptozy indukowanej trójtlenkiem arsenu dużą rolę odgrywają dwa mechanizmy, które powodują zwiększenie ilości białek proapoptotycznych. Pierwszy z nich związany jest z funkcjonowaniem czynnika transkrypcyjnego NF-κB (ang. nuclear factor kappa-light-chain-enhancer of activated B cells). Występuje on w cytoplazmie w stanie nieaktywnym, w kompleksie ze specyficznym inhibitorem IκB(IKK). Składa się on z dwóch podjednostek katalitycznych – IKKα i IKKβ oraz z jednostki regulatorowej IKKγ/NEMO. Fosforylacja inhibitora i jego rozpad uwalnia NF-κB, który wędruje do jądra komórkowego i aktywuje geny odpowiedzialne za produkcję białek "życia" (p53, Bcl-2 i inne inhibitory apoptozy). NF-κB chroni również komórki przed apoptotyczną stymulacją z udziałem receptora TNF-α. Trójtlenek arsenu łączy się z cysteiną znajdującą się w pozycji 179 IKKβ, uniemożliwiając tym samym uwolnienie NF-κB[35]. Brak tego białka w cytoplazmie umożliwia indukcję apoptozy drogą zewnętrzną oraz aktywuje kaspazy 3 i 8[36].
Mechanizm taki zaobserwowano nie tylko w komórkach ostrej białaczki promielocytowej i ziarnicy złośliwej, ale również u pacjentów z zespołem mielodysplastycznym[35][37][38]. Drugim mechanizmem zwiększającym ilość białek proapoptotycznych jest obniżenie transkrypcji genu bcl-2[39]. Działanie to zaobserwowano w komórkach HL-60 i NB4 białaczek ludzkich[40][41].
W 2003 roku naukowcy japońscy odkryli, że trójtlenek arsenu indukuje apoptozę nie tylko przy pomocy receptora TNF-α. Badania wskazują na to, że lek działa proapoptotycznie również poprzez receptor CD95, który wpływa na aktywację kaspaz 8 i 3[42][43]. W komórkach szpiczaka trójtlenek arsenu oddziałuje z receptorem APO2/TRAIL, aktywując kaspazy 8 i 9[44][45].
Trójtlenek arsenu wpływa również na wewnątrzkomórkowe stężenie glutationu, który jest bardzo ważnym elementem układu oksyredukcyjnego (m.in. usuwa wolne rodniki, redukuje nadtlenek wodoru). Bierze on również udział, razem z peroksydazą i katalazą[46], w regulacji poziomu reaktywnych form tlenu. Trójtlenek arsenu hamuje peroksydazę glutationu, zmniejszając tym samym jego stężenie w komórce, co prowadzi do wzrostu poziomu wolnych rodników tlenowych[47]. Te z kolei zwiększają przepuszczalność błony mitochondrium, powodując wypływ białek śmierci i rozpoczęcie procesu apoptozy[48].
Trójtlenek arsenu wpływa również degradująco na polimerazę poli(ADP-rybozy), co w połączeniu z aktywacją kaspaz hamuje naprawę DNA i zatrzymuje cykl komórkowy[49]. To, w jakiej fazie cykl komórkowy zostanie zablokowany, zależy w głównej mierze od białka p53. W komórkach zawierających tzw. typ dziki (niezmutowany) p53 cykl ulega zahamowaniu w interfazie, natomiast w komórkach ze zmutowanym p53 – w fazie G2/M[44][50].
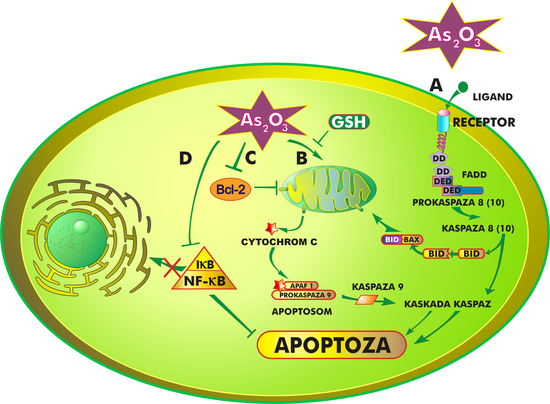
Badania kliniczne
Trójtlenek arsenu był badany klinicznie w dwóch otwartych, jednoramiennych badaniach bez grupy kontrolnej, na 52 pacjentach z ostrą białaczką promielocytową, u których wcześniej bez powodzenia stosowano antracykliny i retinoidy. Wyniki tych badań przedstawia poniższa tabela.
| Badanie jednoośrodkowe n=12 | Badanie wieloośrodkowe n=40 | ||
|---|---|---|---|
| Dawka trójtlenku arsenu (w mg/kg mc./dobę) | 0,16 (mediana: 0,06-0,20) | 0,15 | |
| Całkowita remisja | 11 osób (92%) | 34 osoby (85%) | |
| Średni czas do remisji szpiku kostnego | 32 dni | 35 dni | |
| Średni czas do uzyskania całkowitej remisji | 54 dni | 59 dni | |
| 18-miesięczny okres przeżycia | 67% | 66% | |
| n – liczba pacjentów biorących udział w badaniu | |||
Prowadzono również badania nad działaniem trójtlenku arsenu na inne nowotwory. Wykazały one, że lek działa apoptotycznie również na komórki raka płuc (szczególnie w połączeniu z sulindakiem[55]). Udowodniono również skuteczność trójtlenku arsenu w terapii szpiczaka mnogiego, w połączeniu z kwasem askorbinowym[56] i bortezomibem[57].
Doświadczenia na zwierzętach wykazały, że lek działa również na nowotwory jajnika[58], wątroby, żołądka[59], prostaty, piersi[60] oraz w glejaku[61] i raku trzustki (w połączeniu z partenolidem)[62]. Próby wykorzystania trójtlenku arsenu w leczeniu nowotworów litych zostały ograniczone przez toksyczność leku[63].
Tlenek arsenu wydaje się być obiecującym środkiem w leczeniu chorób autoimmunologicznych (w oparciu o badanie na myszach)[64].
Farmakokinetyka
Nie prowadzono szczegółowych badań odnośnie farmakokinetyki trójtlenku arsenu. Przy podawaniu dożylnym stan równowagi ustala się po 8–10 dniach. Arsen wiąże się z białkami w nieistotnym stopniu. Największe stężenie arsenu wykrywane jest w wątrobie, nerkach, sercu, płucach, włosach i paznokciach. Kwas arsenawy jest utleniany do kwasu arsenowego oraz metylowany w wątrobie[65][66][67], a następnie wydalany w 60% z moczem. Okres półtrwania wynosi 92 godziny. Lek nie jest substratem ani inhibitorem izoenzymów cytochromu P450 (1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1,3A4/5, 4A9/11)[68][69].
Wskazania
Trójtlenek arsenu przeznaczony jest do indukcji remisji i konsolidacji ostrej białaczki promielocytowej (APL, ang. Acute Pro-Myelotic Leucaemia) u dorosłych pacjentów, u których występuje translokacja t(15; 17) i/lub obecność genu PML/RAR α. Lek powinien być stosowany po niepowodzeniu leczenia lub w nawrocie choroby. Wcześniejsza terapia powinna obejmować podawanie retinoidu i chemioterapii[70].
Przeciwwskazania
- Nadwrażliwość na trójtlenek arsenu lub jakikolwiek inny składnik preparatu.
- Ciąża i karmienie piersią są przeciwwskazaniami względnymi (lek przechodzi do mleka matki). Chociaż nie prowadzono badań na kobietach w ciąży, to w badaniach na zwierzętach wykazano teratogenne i embriotoksyczne działanie trójtlenku arsenu. Nie wykazano działania rakotwórczego trójtlenku arsenu, jednakże podobnie jak inne nieorganiczne związki arsenu, jest uznawany za czynnik rakotwórczy.
Ostrzeżenia specjalne
W celu bezpiecznego stosowania leku należy stosować się do poniższych zaleceń[71]:
- U 25% pacjentów leczonych trójtlenkiem arsenu występowały objawy podobne do zespołu aktywacji leukocytów. Charakteryzuje się on wysoką gorączką, dusznościami, przyrostem masy ciała, naciekami płucnymi z obecnością płynu wysiękowego w opłucnej lub osierdziu z leukocytoząlub bez. Stosowanie dużych dawek steroidów (10 mg deksametazonu dożylnie 2–3 razy na dobę) wydaje się łagodzić objawy[72].
- U 40% pacjentów leczonych trójtlenkiem arsenu wystąpił co najmniej raz wydłużony, skorygowany odstęp QT powyżej 500 ms[73]. Wydłużenie odstępu QT może powodować wystąpienie arytmii komorowej typu torsade de pointes[74].
- Przed rozpoczęciem leczenia trójtlenkiem arsenu należy wykonać badanie EKG oraz oznaczyć stężenie potasu, wapnia, magnezu i kreatyniny we krwi. Wszelkie nieprawidłowości powinny zostać skorygowane przed podaniem trójtlenku arsenu, a w szczególności nieprawidłowości w zapisie EKG pod postacią wydłużonego odstępu QT. Jeśli to możliwe, należy przerwać podawanie leków wpływających na wydłużenie odstępu QT[75].
- Pacjenci leczeni trójtlenkiem arsenu, szczególnie ci narażeni na wystąpienie torsade de pointes, powinni być monitorowani podczas podawania leku[76][77].
- Leczenie preparatem należy zmodyfikować lub przerwać przed planowanym zakończeniem leczenia w przypadku, gdy stopień toksyczności osiągnie wartość 3 (według National Cancer Institute). Pacjenci mogą podjąć leczenie z powrotem dopiero po ustąpieniu objawów związanych z toksycznością trójtlenku arsenu. W takich przypadkach należy wznowić leczenie podając 50% wcześniejszej dawki dobowej. Dawkę można zwiększyć do poziomu wcześniejszej dawki dopiero wtedy, gdy w ciągu 3 dni nie wystąpią objawy toksyczności. Pacjenci, u których pojawiły się one po raz drugi, nie mogą kontynuować leczenia trójtlenkiem arsenu.
- U pacjentów w fazie indukcji remisji (patrz: Dawkowanie) należy 2 razy w tygodniu badać stężenie elektrolitów i glukozy we krwi, morfologię oraz czynność wątroby i nerek. Podczas fazy konsolidacyjnej badania te należy wykonywać raz w tygodniu.
- Należy zachować szczególną ostrożność w przypadku pacjentów cierpiących na niewydolność nerek[78].
- Podczas leczenia trójtlenkiem arsenu kobiety w wieku rozrodczym i mężczyźni zdolni do zapłodnienia powinni stosować skuteczne metody antykoncepcji. Nie zbadano dokładnie wpływu trójtlenku arsenu na płodność.
Interakcje
Wiadomo, że trójtlenek arsenu wydłuża odstęp QT. O ile to możliwe, podczas stosowania preparatu nie należy stosować leków wydłużających QT, takich jak[79][80]:
- leki przeciwarytmiczne klasy Ia i III (chinidyna, amiodaron, sotalol, dofetylid)
- niektóre leki stosowane w leczeniu zaburzeń psychicznych (np. amitryptylina, tiorydazyna)
- niektóre makrolidy (np. erytromycyna)
- niektóre leki przeciwhistaminowe (np. terfenadyna, astemizol)
- niektóre chinolony (np. sparfloksacyna)
- inne leki wydłużające odstęp QT (np. cyzapryd)
Stwierdzono również, że wcześniejsze stosowanie:
- antracyklin
- amfoterycyny B
- diuretyków nieoszczędzających potasu
- innych leków powodujących hipokalemię lub hipokalcemię
zwiększa prawdopodobieństwo wystąpienia torsade de pointes.
Działania niepożądane
Działania niepożądane stwierdzono u 37% pacjentów leczonych trójtlenkiem arsenu. Nie miały one jednak dużego nasilenia i mijały w trakcie leczenia. Należy zaznaczyć, że pacjenci znacznie lepiej znoszą leczenie konsolidacyjne niż indukujące remisję. Do najczęściej występujących objawów niepożądanych zaliczyć można:
- hiperglikemię
- hipokalemię
- neutropenię
- zwiększenie aktywności AlAt
- leukocytozę[82][83]
Bardzo nasilone objawy uboczne są dość rzadkie i obejmują:
- zespół aktywowanych leukocytów
- wydłużenie odstępu QT (podczas badań klinicznych odnotowano tylko jeden przypadek torsade de pointes)
- migotanie lub trzepotanie przedsionków
Pozostałe działania niepożądane obejmują skórne reakcje alergiczne (w tym reakcje w miejscu podania, ból w miejscu podania[65]), zaburzenia przewodu pokarmowego (biegunki), bóle różnego rodzaju, zaburzenia widzenia[84], krwawienia[85]. W razie wynaczynienia leku może wystąpić miejscowe podrażnienie i zapalenie żył[65].
Dawkowanie
Schemat stosowania trójtlenku arsenu obejmuje leczenie indukujące remisję, po którym następuje terapia konsolidująca.
W celu indukcji remisji trójtlenek arsenu powinien być podawany we wlewie dożylnym w dawce 0,15 mg/kg mc./dobę codziennie (jednakowo u dzieci, dorosłych i u starszych osób), do momentu uzyskania poprawy w obrazie szpiku kostnego. Leczenie indukujące nie powinno być dłuższe niż 50 dni. W razie braku remisji stwierdzonej w badaniu szpiku kostnego do 50. dnia terapii, należy przerwać leczenie. Leczenie konsolidacyjne (mające na celu eliminację resztkowych komórek nowotworowych) należy rozpocząć w 3–4 tygodniu po zakończeniu leczenia indukującego. Lek podaje się dożylnie w dawce 0,15 mg/kg mc./dobę, 5 dni w tygodniu przez 5 tygodni.
Trójtlenek arsenu powinno podawać się we wlewie trwającym 1–2 godziny. W przypadku wystąpienia reakcji naczynioruchowych czas podawania można wydłużyć do 4 godzin[1].
Przedawkowanie
W przypadku wystąpienia objawów zatrucia arsenem (drgawki, osłabienie mięśniowe, splątanie[86]) należy natychmiast zaprzestać podawania leku oraz wdrożyć odpowiednie leczenie. Najczęściej stosuje się penicylaminę w dawce do 1 g/dobę[87]. U pacjentów, którzy nie mogą przyjmować leków doustnie, można podawać dimerkaprol domięśniowo w dawce 3 mg/kg mc. co 4 godziny[88], aż do ustąpienia objawów zagrażających życiu. W przypadku wystąpienia koagulopatii[89] zaleca się podawanie DMSA w dawce 10 mg/kg mc. co 8 godzin przez 5 dni, a następnie co 12 godzin przez 2 tygodnie[90]. Można również rozważyć zastosowanie dializy[91].
Preparaty i postać leku
- Trisenox – Almac Pharma – 1 mg/ml, koncentrat do sporządzania roztworu do infuzji.
- Trisenox jest pakowany w ampułki o pojemności 10 ml, przeznaczone do jednokrotnego użytku[92]. Ampułki zawierają czysty roztwór trójtlenku arsenu, bez konserwantów, a także wodorotlenek sodu i kwas solny[92]. pH roztworu wynosi 7–9[92]. Lek należy przechowywać w temperaturze pokojowej, nie zamrażać[86]. Zaraz po pobraniu z ampułki lek rozcieńcza się 100–250 ml glukozy 5% lub roztworem soli fizjologicznej. Trójtlenku arsenu nie należy mieszać, ani podawać w jednym wlewie z innymi lekami.
- W preparatyce recepturowej trójtlenek arsenu był stosowany w postaci rozcierki 1:10 z laktozą (Trituratio Acidi arsenicosi 1/10). W celu sporządzenia rozcierki w moździerzu umieszcza się jedną część trójtlenku arsenu i stale ucierając dodaje się porcjami 9 cz. laktozy. Otrzymanie równomiernie rozmieszanej rozcierki wymaga co najmniej 10-minutowego ucierania w moździerzu[2].
---------------
(...) Poza medycyną arsen znalazł zastosowanie w produkcji półprzewodników (jako arsenek galu), polepszania jakości niektórych stopów (m.in. 0,5-2% dodatek do ołowiu poprawia sferyczność amunicji ołowianej, a 0,1-3% dodatek do stopów łożyskowych zawierających ołów zwiększa ich wytrzymałość), w produkcji bojowych środków trujących (luizyty), przy impregnacji drewna i jako dodatek do szkła (dając mu zielonkawą poświatę).









 |
| Źródło: Wikipedia (EN) - Julius Nieuwland. |

*Arseniany (nazwa systematyczna: tetraoksydoarseniany(3−); w systemie Stocka: arseniany(V)) – sole i estry kwasu arsenowego.
Sole kwasu arsenowego zawierać mogą jony AsO3−
4 (arseniany), HAsO2−
4 (wodoroarseniany) lub H
2AsO−
4 (diwodoroarseniany). Są to przeważnie bezbarwne substancje słabo rozpuszczające się w wodzie otrzymywane w reakcjach zobojętniania kwasu arsenowego lub stapiania tritlenku diarsenu.
4 (arseniany), HAsO2−
4 (wodoroarseniany) lub H
2AsO−
4 (diwodoroarseniany). Są to przeważnie bezbarwne substancje słabo rozpuszczające się w wodzie otrzymywane w reakcjach zobojętniania kwasu arsenowego lub stapiania tritlenku diarsenu.
 |
| Źródło: Wikipedia. Garbowanie - grafika z XV wieku. |
*Garbowanie – proces technologiczny prowadzący do zabezpieczenia przed procesami gnilnymi skórysurowej, tzw. golizny i wzrostu odporności termicznej.
Proces garbowania:
Skórę ściągniętą ze zwierzęcia rozwieszano do wyschnięcia (tylko gdy nie można było od razu przystąpić do garbowania), następnie moczono ok. 2 doby w wodzie, po czym usuwano z wewnętrznej strony skóry resztki tkanek podskórnych (tzw. mizdrowanie). Skóry płukano i rozwieszano do obcieknięcia.
Przed przystąpieniem do właściwego garbowania, ściągniętą ze zwierzęcia skórę należy poddać zabiegom wstępnym (tzw. warsztatowi mokremu), mającym na celu usunięcie włosów i rozluźnieniu tkanki skórnej. W tym celu na skórę umieszczoną w dołach wyłożonych połówkami belek i wylepionych gliną, działano substancjami o charakterze zasadowym. Najczęściej stosowano popiół drzewny lub wapno gaszone (np. w Nowym Targu odkryto dół z 2. połowy XII wieku ze skórami przemieszanymi z iłem, popiołem i wapnem).
 Przed garbowaniem usuwano resztki zasad za pomocą kwasowych kąpieli z otrąb (fermentacja) oraz wytrawiano nawozem (kurzym lub gołębim). Zawarte w nim enzymy proteolityczne hydrolizowały białko skórne, zmydlały tłuszcz, usuwały wapno. Skóra po zabiegu była bardziej ciągliwa i miękka. W czasach wcześniejszych w celu usunięcia włosia stosowano tzw. pocenie skóry. Jest to metoda polegająca na celowym dopuszczeniu do zapoczątkowania procesów gnilnych, przy których następuje rozluźnienie nasady włosa i jego wychodzenie, podczas gdy włókna tkanki skórnej pozostawały jeszcze nienaruszone. Metoda ta stosowana jest do dzisiaj przy skórach zwartych (siodlarstwo, futrzarstwo). Skóra ta przy wysokiej wodoodporności jest bardzo wytrzymała na rozciąganie. Można ją zabezpieczyć metodami zarówno fizycznymi, jak i chemicznymi.
Przed garbowaniem usuwano resztki zasad za pomocą kwasowych kąpieli z otrąb (fermentacja) oraz wytrawiano nawozem (kurzym lub gołębim). Zawarte w nim enzymy proteolityczne hydrolizowały białko skórne, zmydlały tłuszcz, usuwały wapno. Skóra po zabiegu była bardziej ciągliwa i miękka. W czasach wcześniejszych w celu usunięcia włosia stosowano tzw. pocenie skóry. Jest to metoda polegająca na celowym dopuszczeniu do zapoczątkowania procesów gnilnych, przy których następuje rozluźnienie nasady włosa i jego wychodzenie, podczas gdy włókna tkanki skórnej pozostawały jeszcze nienaruszone. Metoda ta stosowana jest do dzisiaj przy skórach zwartych (siodlarstwo, futrzarstwo). Skóra ta przy wysokiej wodoodporności jest bardzo wytrzymała na rozciąganie. Można ją zabezpieczyć metodami zarówno fizycznymi, jak i chemicznymi.
Już w neolicie garbowano skóry upolowanych zwierząt poprzez mechaniczne wgniatanie w nie tłuszczu zwierzęcego, tranu lub innych mieszanin. Podczas naciągania skóry i jednoczesnym wcieraniu tłuszczu, wypiera on znajdującą się w niej wodę. Opis tego procesu zachował się w Iliadzie Homera:
„Jak garbarz, namaściwszy tłuszczem skórę wołu, Każe ją mnóstwu ludzi uchwycić pospołu. Ci, gdy równo pociągną, wraz się skóra poci, A tłustość w nią wstępuje na miejsce wilgoci”
Tak potraktowana skóra stawała się miękka, co pozwalało na jej użycie do wyrobu odzieży. Uzyskiwała także zabezpieczenie przed rozkładem i częściową wodoodporność. Metoda ta była stosowana przez tysiąclecia, wykorzystywana była też we wczesnośredniowiecznej Polsce. Indianie w celu nadania skórze pożądanych własności, wędzili ją w dymie jałowcowym. Taka metoda dawała skórę twardą i mniej podatną na przemakanie. Używano także łajna, papki z mózgu i popiołu.
Oprócz garbników mineralnych do garbowania skór stosowano także garbniki roślinne, np. taninę uzyskiwaną z kory dębu, wierzby (do skór delikatnych) i brzozy (do jasnych skór). Garbowano w kadziach, przesypując skóry warstwami zmielonej kory (kadzie z ok. XIII wieku znaleziono w Nysie i Szczecinie). Skóry przekładano z kadzi do kadzi i moczono w brzeczkach (kąpielach garbarskich) o wzrastającym stężeniu. Garbniki roślinne dzieli się na 2 grupy: hydrolizujące i nie hydrolizujące (skondensowane).
*Arsen-76: Nietrwały syntetyczny izotop aresenu o największym zastosowaniu, np. w metodzie atomów znaczonych. Narząd krytyczny stanowi przewód pokarmowy, a dopuszczalne skażenie to 740 kBq.
- Metoda atomów znaczonych – jądrowa metoda analityczna, która wykorzystuje atomy znaczone.Do badanej próbki dodaje się związek chemiczny, który zawiera atomy znaczone, czyli izotop promieniotwórczy. Następnie inicjuje się reakcję i bada się, gdzie po zakończeniu reakcji obserwuje się promieniotwórczość w osadzie, czy w roztworze. Można też celowo dodać izotopy niepromieniotwórcze i śledzić dalej ich rozmieszczenie w próbce. Metoda ta zezwala na badanie mechanizmów reakcji chemicznych, także dla bardzo krótko żyjących izotopów, takich które mają krótki czas połowicznego rozpadu (np. transuranowce).

















ZWIĄZKI ARSENU:
























--> Arseniany (minerały):




























--> Arsenki:



















--> Tlenowe kwasy arsenu:








---> Pochodne tlenowych kwasów arsenu:











--> Organiczne związki arsenu:
a) Heterocykliczne związki arsenu:









b) Związki arsenoorganiczne:


 4) Antymon (Sb, łac. Stibium) - jest to pierwiastek chemiczny z grupy metaloidów. Występuje w czterech odmianach alotropowych: antymon żółty, srebrzystobialy antymon metaliczny, antymon czarny i antymon wybuchowy. Znany jest od starożytności.
4) Antymon (Sb, łac. Stibium) - jest to pierwiastek chemiczny z grupy metaloidów. Występuje w czterech odmianach alotropowych: antymon żółty, srebrzystobialy antymon metaliczny, antymon czarny i antymon wybuchowy. Znany jest od starożytności.
Charakterystyka: Antymon nie reaguje z kwasaminieutleniającymi, gdyż potencjał standardowy Sb(III)/Sb wynosi 0,15 V. Gorący stężony kwas siarkowywprowadza antymon do roztworu w postaci jonów Sb3+, a gorący stężony kwas azotowyprzeprowadza antymon w biały i trudno rozpuszczalny kwas antymonowy. Antymon występuje w roztworze w postaci jonów Sb3+, SbO2−(antymoniny) oraz SbO3− (antymoniany). Antymon tworzy trzy tlenki: Sb
III2O 3, SbV2O 5 oraz mieszany SbIII SbV O 4[5]. Antymonyl (−Sb=O) tworzy rozpuszczalny kompleks z kwasem winowym, winian antymonylu i potasu K(SbO)C4H4O6, znany jako środek wymiotny „emetyk” (łac. emeticus – wymioty).
Izotop 125Sb emituje cząstki beta o energiach od 94 do 621 keV oraz promienie gamma o energiach od 35 do 671 keV. 125Sb najłatwiej gromadzi się w płucach i kościach. Dopuszczalna aktywność w organizmie wynosi 1,5 MBq.
Występowanie: Przybliżona zawartość antymonu w skorupie ziemskiej wynosi od 0,2 do 0,5 ppm (ok. 0,000023% wagowo). Rudami antymonu są antymonit (Sb2S3) i ulmanit (NiSbS).


Zastosowania
- stop drukarski
- stop łożyskowy
- zapałki
- w domieszkowaniu półprzewodników – na przykład InSb (antymonek indu) tworzy półprzewodniki typu „n”
- utwardzacz w stopach ołowiu
- dawniej do wywoływania biegunki jako tzw. pigułka wieczysta – mała kuleczka odlana z antymonu, która po połknięciu powodowała „gwałtowne przeczyszczenie wnętrzności”, po czym mogła być użyta ponownie.

*Babbit – stop łożyskowy z zawartością 83–88% cyny, 8–10% antymonu, 3–6% miedzi i 0,5% ołowiu. Stop ten jest używany na silnie obciążonych panewkach łożysk ślizgowych, np. w silnikach lotniczych i samochodowych. Opracowany w 1839 roku przez Isaaca Babbitta w Taunton, Massachusetts, USA.
*Britannia (metal britannia, początkowo także biały metal) – stop metali zawierający 92% cyny, 6% antymonu i 2% miedzi, po raz pierwszy uzyskany w Sheffield w drugiej połowie XVIII wieku.

ZWIĄZKI ANTYMONU:































5) Bizmut (Bi, łac. Bisemutum, Bismuthum lub Bismutum) - jest to pierwiastek chemiczny, metalbloku p układu okresowego. Nazwa pochodzi od niemieckiego słowa Wismut.















































Właściwości: Czysty bizmut jest kruchym metalem o srebrnym połysku z różowymi refleksami. Jako jedna z nielicznych substancji wykazuje inwersję rozszerzalności termicznej – przy obniżaniu temperatury zmniejsza się jego gęstość, gęstość bizmutu w stanie stałym jest mniejsza niż w stanie ciekłym (podobne właściwości wykazuje woda w temperaturze < 4 °C). Nie reaguje z tlenem i wodą w warunkach normalnych. Roztwarza się w stężonym kwasie azotowym.
Bizmut ogrzany do temperatury topnienia, a następnie wolno oziębiany spontanicznie tworzy kryształy lejkowate; jeśli szybkość oziębiania jest niewielka, rozmiary kryształów mogą być bardzo duże.
Występowanie: Występuje w skorupie ziemskiej w ilości 0,048 ppm (2 razy więcej niż złoto) w postaci trzech rud: bizmutynu (Bi2S3), bizmutytu((BiO)2CO3) i ochry bizmutowej, które stanowią zwykle zanieczyszczenie rud ołowiu i miedzi. Rzadko występuje w postaci rodzimej (elementarnej).



(...)
ZWIĄZKI BIZMUTU: W związkach bizmut jest zazwyczaj trójwartościowy (stopień utlenienia III) i wykazuje właściwości zasadowe. Tworzy tlenek bizmutu(III) Bi2O3, wodorotlenek bizmutu(III) Bi(OH)3 oraz szereg soli zasadowych zawierających ugrupowanie bizmutylowe O=Bi+ (np. chlorek bizmutylu, O=Bi-Cl). Jako jedyny pierwiastek 15 grupy tworzy trwałe sole z kwasami tlenowymi, np. siarczan bizmutu(III), Bi2(SO4)3. Ponadto znane są sole bizmutu i kwasów beztlenowych(halogenki BiX3, siarczek bizmutu(III) Bi2S3). Wszystkie sole bizmutu łatwo ulegają hydrolizie do soli bizmutylowych.

(...) Bizmutowodór (bizmutyna), BiH3, jest nietrwałym, trującym gazem (temp. wrz. ok. 20 °C) o właściwościach redukujących. Bizmut(III) tworzy też bezpośrednie połączenia z metalami, bizmutki typu M3Bi, np. bizmutek sodu Na3Bi i bizmutek magnezu Mg3Bi2.
 |
| Źródło: Wikipedia. Syntetyczne kryształy czystego bizmutu. Obok sześcian (1 cm³) wykonany z bizmutu o czystości 99,99%. |
Znanych jest wiele związków kompleksowychbizmutu, np. zawierających anion [BiCl4]-, [BiCl5]2-, [BiCl5]3-, [Bi2Cl7]-, [Bi(SO4)2]- i in. Z ligandami kleszczowymi tworzy chelaty, np. [Bi(O2C6H4)2]-. Halogenki bizmutu są kwasami Lewisa i z donorami elektronów tworzą kompleksy typu Et2O→BiCl3.
- Ligandy chelatujące (kleszczowe) – ligandy w cząsteczkach, których wiązania koordynacyjne zostały utworzone pomiędzy atomem centralnym a dwoma lub więcej atomami należącymi do cząsteczki ligandu.
 |
| Źródło: Wikipedia. Lejkowaty kryształ bizmutu. |
Na stopniu utlenienia V bizmut wykazuje właściwości kwasowe i tworzy nietrwałe sole – bizmutany typu MBiO3 o silnych właściwościach utleniających (np. bizmutan potasowy, KBiO3).
- Związki bizmutoorganiczne
Podobnie jak pozostałe pierwiastki grupy 15, bizmut(III) tworzy połączenia z resztami organicznymi typu R3Bi oraz R3BiZ2 (R – reszta organiczna, Z – anion nieorganiczny), np. (CH3)3Bi, Ph3Bi, Ph3BiF2 lub Ph3Bi(OH)2.
Izotopy: Bizmut ma 35 izotopów z przedziału mas 184–218. Żaden z nich nie jest trwały. W 2003 roku we francuskim Institut d'Astrophysique Spatiale w Orsay wyznaczono dokładnie półokres rozpadu najtrwalszego izotopu bizmutu (209Bi), który wynosi ok. 1,9×1019 lat (tj. ponad miliard razy więcej niż szacowany wiek Wszechświata), wcześniej szacowanego na 1018 lat. Ta śladowa radioaktywność nie stanowi żadnego zagrożenia biologicznego, ma jednak znaczenie naukowe, gdyż potwierdziła wcześniejsze obliczenia teoretyczne wskazujące na niestabilność wszystkich izotopów bizmutu. W naturalnym bizmucie występują też śladowe ilości radioizotopów, np. 210Bi (ok. 50 ppm składu izotopowego).
Bizmut-210: Emituje promieniowanie beta o energii 1,162 MeV, przekształcając się w 210Po. Często występuje w równowadze promieniotwórczej ze swoim prekursorem, 210Pb. Jest wysoce radiotoksyczny. Narząd krytyczny stanowią nerki, a dopuszczalne skażenie zostało ustalone na 1,5 kBq.

Znaczenie biologiczne bizmutu: Znaczenie biologiczne – brak lub nieznane. Występuje w kościach i krwi (ok. 0,2 ppm). Jego sole i tlenki są nietoksyczne, mimo że jest metalem ciężkim. Sole bizmutu stosowane są w leczeniu wrzodów żołądka spowodowanych zakażeniem Helicobacter pylori. Niewiele wiadomo o toksyczności bizmutu. Nie wykazano upośledzenia i odstępstw od normy w rozwoju szczurów, którym przez 28 dni podawano bizmut w dawkach 0, 40, 200,1000 mg na kg masy ciała, dla obu płci; ustalono, iż LD50 > 2000 mg/kg masy ciała (vide: dawka śmiertelna).


ZWIĄZKI BIZMUTU:

--> Organiczne związki bizmutu:
a) Heterocykliczne związki bizmutu:




b) Organiczne sole bizmutu:


--> Sole bizmutu:
a) Nieorganiczne sole bizmutu:






























b) Organiczne sole bizmutu:
- Jodek bizmutawo-chininowy,
- Kseroform

6) Moscovium (Mc) - jest to syntetyczny pierwiastek chemiczny, transuranowiec o liczbie atomowej 115. W układzie okresowym znajduje się w bloku p bezpośrednio pod bizmutem i bywał nazywany nieoficjalnie eka-bizmutem.

Brak komentarzy:
Prześlij komentarz