
 Adenozyna - jest to związek chemiczny z grupy nukleozydów, który zbudowany jest z adeniny połączonej z pierwszym węglem pierścienia rybozy wiązaniem beta-N9-glikozydowym. Adenozyna odgrywa ważną rolę w wielu procesach biochemicznych. Odpowiada za transport energii jako trifosforan adenozyny (ATP), a także difosforan adenozyny (ADP); przekazuje informację genetyczną, stanowiąc składnik RNA, przekazuje informację w transdukcji sygnału jako wtórny przekaźnik cAMP; działa jako neuroprzekaźnik hamujący w ośrodkowym układzie nerwowym (OUN); a także uczestniczy w reakcjach metylacji jako S-adenozylometionina. Jako metabolit ATP - reguluje ona również średnicę naczyń, powoduje skurcz naczyń nerkowych i rozkurcz pozostałych łożysk naczyniowych. Jest ona również istotnym metabolitem w autoregulacji krążenia mózgowego i przepływu wrotnego przez wątrobę.
Adenozyna - jest to związek chemiczny z grupy nukleozydów, który zbudowany jest z adeniny połączonej z pierwszym węglem pierścienia rybozy wiązaniem beta-N9-glikozydowym. Adenozyna odgrywa ważną rolę w wielu procesach biochemicznych. Odpowiada za transport energii jako trifosforan adenozyny (ATP), a także difosforan adenozyny (ADP); przekazuje informację genetyczną, stanowiąc składnik RNA, przekazuje informację w transdukcji sygnału jako wtórny przekaźnik cAMP; działa jako neuroprzekaźnik hamujący w ośrodkowym układzie nerwowym (OUN); a także uczestniczy w reakcjach metylacji jako S-adenozylometionina. Jako metabolit ATP - reguluje ona również średnicę naczyń, powoduje skurcz naczyń nerkowych i rozkurcz pozostałych łożysk naczyniowych. Jest ona również istotnym metabolitem w autoregulacji krążenia mózgowego i przepływu wrotnego przez wątrobę. Anandamid (AEA) - stanowi organiczny związek chemiczny z grupy psychoaktywnych kannabinoidów (związków, które po przyłączeniu się do receptorów kannabinoidowych komórki blokują wpływ jonów wapnia z płynu zewnątrzkomórkowego do cytoplazmy oraz hamują wytwarzanie cAMP. W wyniku tego zmienia się metabolizm komórek i dochodzi do zmian w uwalnianiu hormonów a także neuroprzekaźników) występującym w organizmach żywych.
Anandamid (AEA) - stanowi organiczny związek chemiczny z grupy psychoaktywnych kannabinoidów (związków, które po przyłączeniu się do receptorów kannabinoidowych komórki blokują wpływ jonów wapnia z płynu zewnątrzkomórkowego do cytoplazmy oraz hamują wytwarzanie cAMP. W wyniku tego zmienia się metabolizm komórek i dochodzi do zmian w uwalnianiu hormonów a także neuroprzekaźników) występującym w organizmach żywych.2 - Arachidonyloglicerol (2-AG) - jest związkiem organicznym z grupy endokannabinoidów i agonistą receptora CB1. 2-AG jest estrem kwasu tłuszczowego omega-6 - kwasu arachidonowego i glicerolu.

Endorfiny - endorfiny to grupa hormonów peptydowych, które wywołują dobre samopoczucie i zadowolenie z siebie a także generalnie wywołują wszelkiego rodzaju inne stany euforyczne; tłumią one również odczuwanie drętwienia i bólu. Są ponadto endogennymi opioidami. Stanowią silne agonisty receptorów opioidowych μ, których pobudzanie wywołuje stany euforyczne. Na te same receptory działają opioidy egzogenne, co wywołuje zniesienie bólu, uczucie przyjemności, a także dobrego nastroju. Wywołuje to jednakże również silne uzależnienie psychiczne i fizyczne.
John Hughes i Hans Kosterlitz podali chemiczny skład tajemniczej substancji nazwanej później endorfiną (wewnętrzną morfiną). Wytwarzana przez mózg, działa podobnie jak morfina, zmniejsza ból i wprawia w błogostan. Wkrótce odkryto kilkanaście różnych endorfin produkowanych w mózgu i rdzeniu kręgowym, które wpływają na odczuwanie bólu, stan uczuć i świadomości. Nazwano je molekułami emocji. Najwięcej receptorów emocji znaleziono w mózgu, w obszarach uważanych za siedzibę uczuć i ośrodki odczuwania przyjemności. W latach 60. w eksperymentach na szczurach pokazano, że elektryczna stymulacja tychże miejsc budzi w zwierzętach doznania tak intensywnej ekstazy, że wolą paść z wyczerpania aniżeli przerwać doświadczenia. Wydzielanie endrofin spowodować może wiele bodźców, m.in. śmiech (lub nawet myślenie o śmiechu), wysiłek fizyczny (ew. udział w rywalizacji sportowej), przedłużony intensywny wysiłek zaś powoduje wzmożone wydzielanie endorfin, które objawia się tzw. euforią biegacza, który powoduje pojawienie się u człowieka zwiększonej odporności na ból oraz zmęczenie. Moment wystąpienia efektu odpowiada czasowi, po którym mięśnie zużywają cały glikogen w nich zmagazynowany. Po upływie ok. 45-60 minut podczas wysiłku następuje moment przejścia z oddychania aerobowego na anaerobowe (tzw. próg anerobowy), kiedy to powstaje tzw. dług tlenowy. Niedotlenienie wywołuje stres organizmu, co z kolei może powodować silne wydzielanie endorfin. Podczas krótszych, intensywnych ćwiczeń również są wydzielane endorfiny, jednakże w ilościach niemierzalnych; uwolnienie endorfin przygotowuje zaś organizm na dalszy wysiłek fizyczny. Produkcja endorfin wywołuje emocjonalną reakcję na stres, która objawia się poprawą nastroju i zwiększeniem wytrzymałości i odporności organizmu na ból, co umożliwia dalszy wysiłek, który w normalnych warunkach nie mógłby być kontynuowany. Biegacze opisują to jako przejęcie przez umysł kontroli nad ciałem. Znane są jednakże również przypadki negatywnych odczuć u osób ćwiczących, gdzie zamiast poprawy nastroju, obserwuje się jego obniżenie za sprawą negatywnego wpływu warunków zewnętrznych, np. podczas biegania w zimny deszczowy dzień po zmroku, w nieznanych terenie lub podczas ppływania w zimnej wodzie. Sama teoria euforii biegacza zrodziła się w latach 70-tych w Stanach Zjednoczonych, kiedy odkryte zostały receptory opioidowe μ (my) w mózgu. Endorfiny bowiem silnie oddziałują na owe receptory (są zatem ich agonistami), których pobudzanie wywołuje z kolei stany euforyczne (stany wyjątkowo dobrego samopoczucia w których występuje tendencja do śmiechu, radości, płytkiej wesołości i dowcipkowania). Aktywność fizyczna, podczas której może wystąpić stan euforyczny obejmuje nie tylko bieganie, lecz również pływanie, narciarstwo biegowe, wioślarstwo długodystansowe, jazdę na rowerze, podnoszenie ciężarów, kulturystykę, aerobik, gry zespołowe takie jak koszykówka lub piłka nożna. Ciekawostką jest to, iż niektórzy fizjologowie twierdzą, iż biegacze długodystansowi to "endogenni morfiniści", którzy są uzależenieni fizjologicznie od biegania i to właśnie maratończycy są grupą gdzie fenomen euforii występuje najczęściej. Sugeruje się, że może on stanowić główną przyczynę uprawiania biegó długodystansowych. Znane są również przypadku uzależnienia od wysiłku fizycznego, które powodują szkody fizyczne i psychiczne. Wśród himalaistów natomiast lub alpinistów, występuje efekt euforii wysokościowej, do której dochodzi na skutek niedotlenienia mózgu na dużej wysokości. Niepotwierdzona jeszcze teoria mówi, iż niedobór tlenu podczas wspinaczki wywołuje stres organizmu, który w odpowiedzi na niego wytwarza w płynie mózgowo-rdzeniowym endorfiny. Himalaiści opisują przypadki euforii wśród wspinaczy, które objawiają się porzucaniem elementów sprzętu i ubioru, a nawet halucynacjami. Badania nad wpływem endorfin na człowieka nie należą do łatwych przedsięwzięć. Ogromna liczba naukowych doniesień o wpływie endorfin na system opioidowy tak naprawdę pochodzi z inwazyjnych badań przeprowadzanych na szczurach i myszach, u których bada się bezpośrednio zmiany mózgu. Niestety oznacza to zazwyczaj śmierć obiektu badań. Badania na szczurach dowiodły, iż intensywny długotrwały wysiłek powoduje wzrost poziomu endorfin w mózgu, co z kolei indukuje zwiększony próg odczuwania bólu. Wysiłek wpływa także na mechanizm oddziaływania dynorfin i enkefalin - pozostałych dwóch grup endogennych peptydów opioidowych, na receptory opioidowe. U ludzi natomiast badania prowadzi się w sposób dwojaki. Typowe badania polegają na oznaczaniu zawartości endorfin i innych substancji we krwi, przy założeniu, że jest ona powiązana z poziomem w mózgu (jednakże tak naprawdę transport endorfin pomiędzy mózgiem i krwią nie zachodzi łatwo). Drugi sposób polega na obserwacji zmian jakie występują u ochotników, którym podaje się blokery receptorów opioidowych μ (my). Badania prowadzone u ludzi potwierdziły, że poziom endorfin zależy od intensywności ćwiczeń: przy wysiłku aerobowym o dużej intensywności jest on większy niż w stanie spoczynku, natomiast przy łagodnym wysiłku aerobowym poziom endorfin nie zmienia się w porównaniu do stanu spoczynku. Sama euforia biegacza jest zjawiskiem dużo bardziej złożonym i związanym również z innymi aniżeli endorfiny substancjami. Tu mowa jednak o endorfinach. Bodźcami, które powodują ich wydzielanie są: śmiech (a nawet samo myślenie na temat śmiechu); wysiłek fizyczny; niedotlenienie - istnieje bowiem hipoteza działania euforyzującego alkoholu na organizm poprzez zwiększone powinowactwo tej substancji do tlenu; niektóre przyprawy, np. papryka chili (u szczurów); w niektórych przypadkach akupunktura; opalanie, czekolada, niektóre psychoaktywne substancje i orgazm.
Glicyna (Acidum aminoaceticum) - glicyna posiada najmniejszą resztę aminokwasową, z jednym tylko atomem wodoru w łańcuchu bocznym. Ze względu na to, że z atomem węgla alfa związane są dwa atomy wodoru, glicyna - w przeciwieństwie do innych aminokwasów - nie jest optycznie czynna. Glicyna zalicza się do grupy aminokwasów niepolarnych alifatycznych. W trakcie ewolucji dywergentnej (rozbieżnej) reszty glicyny zmieniają się znacznie rzadziej niż pozostałych aminokwasów, a gdy już ulegają mutacji w białkach homologicznych, to na takie reszty jak alanina, seryna, kwas asparaginowy lub asparagina. Glicyna jako samodzielny aminokwas występuje przede wszystkim w roli przekaźnika w ośrodkowym układzie nerwowym. Działa jako hamujący przekaźnik receptorów glicynowych, a ponadto jako koagonista kluczowy do aktywacji receptorów NMDA (receptorów N-metylo-D-asparaginowych - rodzajów receptora dla glutaminianu, który jest selektywnie aktywowany przez kwas N-metylo-D-asparaginowy (NMDA). Jest to receptor jonotropowy, który przewodzi kationy: sodu (Na+), potasu (K+) i wapnia (Ca2+). Do aktywacji glutaminianu wymagania przyłączenia glicyny lub seryny; dodatkowym warunkiem aktywacji receptora NMDA jest depolaryzacja komórki, ponieważ przy potencjale spoczynkowym jest on blokowany przez jon magnezu (Mg2+)). Stężenie glicyny w OUN jest wyższe w rdzeniu przedłużonym, moście oraz rdzeniu kręgowym, natomiast niższe w półkulach mózgowych i móżdżku. Inną fizjologiczną funkcją glicyny jest jej obecności w peroksysomach hepatocytów - wielobocznych komórek wątrobowych, które stanowią podstawowy element strukturalny miąższu wątroby. Glicyna ulega tam sprzęgnięciu z pierwotnymi kwasami żółciowymi i tworzy w ten sposób sole żółciowe. Glicyna bierze ponadto udział w biosyntezie puryn de novo, w trakcie której wbudowana zostaje do pierścienia nukleotydowego, będąc źródłem węgli C4 i C5 a także azotu N7 w owym pierścieniu. Buduje więc ona pierścień imidazolowy. Przemiana ta wygląda w sposób następujący:

John Hughes i Hans Kosterlitz podali chemiczny skład tajemniczej substancji nazwanej później endorfiną (wewnętrzną morfiną). Wytwarzana przez mózg, działa podobnie jak morfina, zmniejsza ból i wprawia w błogostan. Wkrótce odkryto kilkanaście różnych endorfin produkowanych w mózgu i rdzeniu kręgowym, które wpływają na odczuwanie bólu, stan uczuć i świadomości. Nazwano je molekułami emocji. Najwięcej receptorów emocji znaleziono w mózgu, w obszarach uważanych za siedzibę uczuć i ośrodki odczuwania przyjemności. W latach 60. w eksperymentach na szczurach pokazano, że elektryczna stymulacja tychże miejsc budzi w zwierzętach doznania tak intensywnej ekstazy, że wolą paść z wyczerpania aniżeli przerwać doświadczenia. Wydzielanie endrofin spowodować może wiele bodźców, m.in. śmiech (lub nawet myślenie o śmiechu), wysiłek fizyczny (ew. udział w rywalizacji sportowej), przedłużony intensywny wysiłek zaś powoduje wzmożone wydzielanie endorfin, które objawia się tzw. euforią biegacza, który powoduje pojawienie się u człowieka zwiększonej odporności na ból oraz zmęczenie. Moment wystąpienia efektu odpowiada czasowi, po którym mięśnie zużywają cały glikogen w nich zmagazynowany. Po upływie ok. 45-60 minut podczas wysiłku następuje moment przejścia z oddychania aerobowego na anaerobowe (tzw. próg anerobowy), kiedy to powstaje tzw. dług tlenowy. Niedotlenienie wywołuje stres organizmu, co z kolei może powodować silne wydzielanie endorfin. Podczas krótszych, intensywnych ćwiczeń również są wydzielane endorfiny, jednakże w ilościach niemierzalnych; uwolnienie endorfin przygotowuje zaś organizm na dalszy wysiłek fizyczny. Produkcja endorfin wywołuje emocjonalną reakcję na stres, która objawia się poprawą nastroju i zwiększeniem wytrzymałości i odporności organizmu na ból, co umożliwia dalszy wysiłek, który w normalnych warunkach nie mógłby być kontynuowany. Biegacze opisują to jako przejęcie przez umysł kontroli nad ciałem. Znane są jednakże również przypadki negatywnych odczuć u osób ćwiczących, gdzie zamiast poprawy nastroju, obserwuje się jego obniżenie za sprawą negatywnego wpływu warunków zewnętrznych, np. podczas biegania w zimny deszczowy dzień po zmroku, w nieznanych terenie lub podczas ppływania w zimnej wodzie. Sama teoria euforii biegacza zrodziła się w latach 70-tych w Stanach Zjednoczonych, kiedy odkryte zostały receptory opioidowe μ (my) w mózgu. Endorfiny bowiem silnie oddziałują na owe receptory (są zatem ich agonistami), których pobudzanie wywołuje z kolei stany euforyczne (stany wyjątkowo dobrego samopoczucia w których występuje tendencja do śmiechu, radości, płytkiej wesołości i dowcipkowania). Aktywność fizyczna, podczas której może wystąpić stan euforyczny obejmuje nie tylko bieganie, lecz również pływanie, narciarstwo biegowe, wioślarstwo długodystansowe, jazdę na rowerze, podnoszenie ciężarów, kulturystykę, aerobik, gry zespołowe takie jak koszykówka lub piłka nożna. Ciekawostką jest to, iż niektórzy fizjologowie twierdzą, iż biegacze długodystansowi to "endogenni morfiniści", którzy są uzależenieni fizjologicznie od biegania i to właśnie maratończycy są grupą gdzie fenomen euforii występuje najczęściej. Sugeruje się, że może on stanowić główną przyczynę uprawiania biegó długodystansowych. Znane są również przypadku uzależnienia od wysiłku fizycznego, które powodują szkody fizyczne i psychiczne. Wśród himalaistów natomiast lub alpinistów, występuje efekt euforii wysokościowej, do której dochodzi na skutek niedotlenienia mózgu na dużej wysokości. Niepotwierdzona jeszcze teoria mówi, iż niedobór tlenu podczas wspinaczki wywołuje stres organizmu, który w odpowiedzi na niego wytwarza w płynie mózgowo-rdzeniowym endorfiny. Himalaiści opisują przypadki euforii wśród wspinaczy, które objawiają się porzucaniem elementów sprzętu i ubioru, a nawet halucynacjami. Badania nad wpływem endorfin na człowieka nie należą do łatwych przedsięwzięć. Ogromna liczba naukowych doniesień o wpływie endorfin na system opioidowy tak naprawdę pochodzi z inwazyjnych badań przeprowadzanych na szczurach i myszach, u których bada się bezpośrednio zmiany mózgu. Niestety oznacza to zazwyczaj śmierć obiektu badań. Badania na szczurach dowiodły, iż intensywny długotrwały wysiłek powoduje wzrost poziomu endorfin w mózgu, co z kolei indukuje zwiększony próg odczuwania bólu. Wysiłek wpływa także na mechanizm oddziaływania dynorfin i enkefalin - pozostałych dwóch grup endogennych peptydów opioidowych, na receptory opioidowe. U ludzi natomiast badania prowadzi się w sposób dwojaki. Typowe badania polegają na oznaczaniu zawartości endorfin i innych substancji we krwi, przy założeniu, że jest ona powiązana z poziomem w mózgu (jednakże tak naprawdę transport endorfin pomiędzy mózgiem i krwią nie zachodzi łatwo). Drugi sposób polega na obserwacji zmian jakie występują u ochotników, którym podaje się blokery receptorów opioidowych μ (my). Badania prowadzone u ludzi potwierdziły, że poziom endorfin zależy od intensywności ćwiczeń: przy wysiłku aerobowym o dużej intensywności jest on większy niż w stanie spoczynku, natomiast przy łagodnym wysiłku aerobowym poziom endorfin nie zmienia się w porównaniu do stanu spoczynku. Sama euforia biegacza jest zjawiskiem dużo bardziej złożonym i związanym również z innymi aniżeli endorfiny substancjami. Tu mowa jednak o endorfinach. Bodźcami, które powodują ich wydzielanie są: śmiech (a nawet samo myślenie na temat śmiechu); wysiłek fizyczny; niedotlenienie - istnieje bowiem hipoteza działania euforyzującego alkoholu na organizm poprzez zwiększone powinowactwo tej substancji do tlenu; niektóre przyprawy, np. papryka chili (u szczurów); w niektórych przypadkach akupunktura; opalanie, czekolada, niektóre psychoaktywne substancje i orgazm.
Glicyna (Acidum aminoaceticum) - glicyna posiada najmniejszą resztę aminokwasową, z jednym tylko atomem wodoru w łańcuchu bocznym. Ze względu na to, że z atomem węgla alfa związane są dwa atomy wodoru, glicyna - w przeciwieństwie do innych aminokwasów - nie jest optycznie czynna. Glicyna zalicza się do grupy aminokwasów niepolarnych alifatycznych. W trakcie ewolucji dywergentnej (rozbieżnej) reszty glicyny zmieniają się znacznie rzadziej niż pozostałych aminokwasów, a gdy już ulegają mutacji w białkach homologicznych, to na takie reszty jak alanina, seryna, kwas asparaginowy lub asparagina. Glicyna jako samodzielny aminokwas występuje przede wszystkim w roli przekaźnika w ośrodkowym układzie nerwowym. Działa jako hamujący przekaźnik receptorów glicynowych, a ponadto jako koagonista kluczowy do aktywacji receptorów NMDA (receptorów N-metylo-D-asparaginowych - rodzajów receptora dla glutaminianu, który jest selektywnie aktywowany przez kwas N-metylo-D-asparaginowy (NMDA). Jest to receptor jonotropowy, który przewodzi kationy: sodu (Na+), potasu (K+) i wapnia (Ca2+). Do aktywacji glutaminianu wymagania przyłączenia glicyny lub seryny; dodatkowym warunkiem aktywacji receptora NMDA jest depolaryzacja komórki, ponieważ przy potencjale spoczynkowym jest on blokowany przez jon magnezu (Mg2+)). Stężenie glicyny w OUN jest wyższe w rdzeniu przedłużonym, moście oraz rdzeniu kręgowym, natomiast niższe w półkulach mózgowych i móżdżku. Inną fizjologiczną funkcją glicyny jest jej obecności w peroksysomach hepatocytów - wielobocznych komórek wątrobowych, które stanowią podstawowy element strukturalny miąższu wątroby. Glicyna ulega tam sprzęgnięciu z pierwotnymi kwasami żółciowymi i tworzy w ten sposób sole żółciowe. Glicyna bierze ponadto udział w biosyntezie puryn de novo, w trakcie której wbudowana zostaje do pierścienia nukleotydowego, będąc źródłem węgli C4 i C5 a także azotu N7 w owym pierścieniu. Buduje więc ona pierścień imidazolowy. Przemiana ta wygląda w sposób następujący:

Ponadto glicyna wraz z sukcynylo-CoA bierze udział w syntezie hemu.
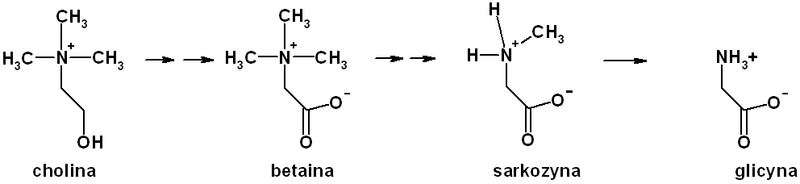
Ponieważ ludzki organizm potrafi syntezować glicynę jest ona nazywana aminokwasem endogennym. Może być ona produkowana: z glioksalanu i glutaminianu przez aminotransferazę glutaminianową; z alaniny przez aminotransferazę alaninową. Ważnym sposobem syntezy glicyny u ssaków jest również synteza z choliny oraz seryny. Poniżej przedstawiony jest uproszczony szlak syntezy glicyny z choliny. Cholina ulega przemianie w betainę. Betaina ulega przemianie w sarkozynę. Ostatecznie przemiany choliny doprowadzają do powstania glicyny.
Glicyna ulega degradacji w organizmie na drodze trzech szlaków metabolicznych. Glicyna może np. ulec przekształceniu w serynę. Reakcję tę katalizuje hydroksymetylotransferaza serynowa, zaś koenzymami reakcji są tetrahydrofolian i fosforan pirydoksalu. Degradacja glicyny może jednakże również zachodzić w wątrobie za pomocą mitochondrialnego kompleksu syntazy glicynowej, który potrafi rozłożyć ją na dwutlenek węgla oraz jon amonowy, przy okazji tworząc N5,N10-metylenotetrahydrofolian. Kofaktorem tego procesu jest fosforan pirydoksalu. Owa ścieżka metaboliczna ma olbrzymie znaczenie dla ssaków, zaś jej zaburzenia są w stanie doprowadzić do hiperglicynemii. Owa ścieżka metaboliczna degradacji glicyny wraz z przedstawieniem kolorami losów poszczególnych grup atomów przedstawiona została poniżej:

Trzecim sposobem degradacji glicyny jest utlenienie przez oksydazę D-aminokwasów. Glicyna zostaje przekształcona do glioksalanu, który z kolei jest utleniany w NAD+-zależnej reakcji do szczawianu. Ponadto glicyna ulega licznym przemianom w inne metabolity, co zostało opisane w podrozdziale dotyczącym jej funkcji.
Chorobami związanymi z przemianami glicyny są schorzenia takie jak, np. Glicynuria, która wynika z zaburzeń reabsorpcji glicyny w kanalikach nerkowych i polega na wydalaniu większych aniżeli normalnie ilości tego aminokwasu. Innym zaburzeniem jest pierwotna huperoksaluria - zaburzenie katabolizmu glioksalanu, który powstaje przez deaminację glicyny. Następujące w nim utlenienie glioksalanu do szczawianu skutkuje kamicą moczową a także wapnicą nerek. Może to doprowadzić do przedwczesnych zgonów na skutek niewydolności nerek a także nadciśnienia. Inną chorobą związaną z przemianami glicyny jest nieketonowa hiperglicynemia - choroba genetyczna, która powoduje nagromadzanie się glicyny we krwi i płynie mózgowo - rdzeniowym.
Histamina (β-imidazolyloetyloamina) - jest to organiczny związek chemiczny, heterocykliczna amina, która stanowi pochodną imidazolu. Zawiera ona boczny łańcuch etyloaminowy. Występuje naturalnie w organizmie ludzkim, gdzie pełni funkcję mediatora procesów zapalnych, mediatora odczynu alergicznego, neuroprzekaźnika, a także pobudza wydzielanie kwasu żołądkowego. Histamina jest hormonem tkankowym (zwierzęcym hormonem uczestniczącym w lokalnym pozanerwowym sterowaniu działaniem narządów) zaliczanym do neurohormonów, wytwarzanym z aminokwasu histydyny. W ludzkim organizmie powstaje w wyniku dekarboksylacji (w obecności fosforanu pirydoksalu) histydyny. Jej produkcja zachodzi w wielu miejscach. Najwyższe stężenia zaobserwować można w płucach, skórze, błonie śluzowej nosa i żołądka. Histamina magazynowana jest w formie nieczynnej w ziarnistościach bazofili - granulocytów zasadochłonnych i mastocytów - komórek tucznych, które znajdują się w tkance łącznej, przede wszystkim w okolicy naczyń krwionośnych i limfatycznych, a także nerwów, skąd może uwalniana być w reakcji zapalnej. W żołądku histamina występuje w histaminocytach, zaś w ośrodkowym układzie nerwowym w neuronach histaminergicznych.
Aminy biogenne (w tym histamina) występują w wielu artykułach żywnościowych, przede wszystkim wytwarzanych i dojrzewających przy udziale procesów fermentacyjnych, a także nieświeżych, lub silnie skażonych mikrobiologicznie. Prekursorami tychże amin są aminokwasy, które uwalniane są z białek na drodze hydrolizy.
Histamina głównie występuje w surowcach rybnych. Spożycie dużej jej ilości jest powiązane z zatruciem pokarmowym. Zawartość histaminy w surowcach i przetworach rybnych zależy przede wszystkim od ilości wolnej histydyny w mięśniach, obecności aktywatorów i inhibitorów dekarboksylaz, a także od rodzajów i wielkości populacji bakteryjnej.
W wyniku reakcji antygenu z przeciwciałami we krwi człowieka lub pod wpływem czynników niszczących błonę komórki tucznej magazynującej histaminę, np wskutek zimna lub ucisku, a także pod wpływem zmian pH, składu jonów oraz leków pobudzających aktywność histaminy) dochodzi do uwalniania histaminy i rozwoju reakcji zapalnej. Histamina wyzwolona z tkanek wiąże się ze swoistymi receptorami, a następnie ulega przemianie do nieczynnych produktów. Po degranulacji komórek tucznych histamina przenika do naczyń krwionośnych, a jej poziom we krwi rośnie między 2,5 a 5 minutami i wraca do poziomu wyjściowego po 15-30 minutach. Działanie histaminy opiera się na pobudzaniu receptorów H1, H2, H3 i H4. Efekty owego pobudzenia są następujące dla poszczególnych receptorów:

Histamina uczestniczy w zapaleniu alergicznym jako mediator prozapalny nie tylko wczesnej, ale i późnej fazy odczynu, ale prawdopodobnie wpływa również na odległe następstwa choroby w postaci przebudowy dróg oddechowych. W reakcji alergicznej natychmiastowej centralną rolę odgrywa proces aktywacji receptora H1. Kontakt z alergenem powoduje wylew histaminy z ziarnistości niektórych ludzkich komórek odpornościowych, np. mastocytów i silne podrażnienie błony śluzowej jelit, nosa, oskrzeli, płuc oraz tkanki skórnej. Odpowiednio do tych lokalizacji wywołuje to: pokrzywkę skórną, biegunkę, katar sienny, astmę, a u niektórych wstrząs anafilaktyczny, np. wskutek ukąszenia osy lub pszczoły. Histamina odpowiada ponadto za wystąpienie objawów:
- alergii wziewnej oraz skórnej - wywołuje bowiem zaczerwienienie, lokalne ocieplenie, nabrzmienie i bolesny stan zapalny. Rozszerza małe naczynia krwionośne i zwiększa ich przepuszczalność; powoduje lokalne wysięki osocza i obrzęki (nosa, gardła, oskrzeli, a także obrzęk naczyniorychowy i obecność bąbli przy pokrzywce). Drażnienie przez histaminę zakończeń nerwów czuciowych powoduje świąd i doznania bólowe.
- astmy - jej nagły wylew grozi bowiem obrzękiem krtani, tchawicy, zaś w najostrzejszych przypadkach obrzękiem płuc, co odcina dopływ powietrza. Zmiany te powodują utrudnienia w oddychaniu, które stanowią główny objaw dychawicy oskrzelowej,
- alergii pokarmowej - jej obecność w jelitach powoduje bowiem skurcz mięśni gładkich i wzmożone wydzielanie soków trawiennych. Podrażniona błona śluzowa jelita cienkiego reaguje stanem zapalnym, który wywołuje biegunkę,
- anafilaksji - po przekroczeniu pewnego progu ilościowego histamina przedostaje się z tkanek do krążenia i może wywołać niebezpieczne reakcje ogólnoustrojowe, które objawiają się nagłym spadkiem ciśnienia krwi oraz przyspieszeniem akcji serca, co może grozić zgonem,
- przewlekłej alergii, np. całorocznego alergicznego nieżytu nosa lub przewlekła pokrzywka idiopatyczna - stale uwalniana histamina bowiem drastycznie zwiększa przepuszczalność ścian naczyń krwionośnych wskutek ciągłego drażnienia tzw. receptorów histaminowych H1. Przewlekły stan zapalny i drażniące działanie histaminy oraz innych mediatorów powodują stopniową degenerację okolicznych tkanek.
Histamina zmienia również właściwości błony komórkowej, wskutek czego do wnętrza komórki dostaje się zbyt dużo jonów wapnia i sodu. Może to wywoływać nadmierne skurcze mięśni oskrzeli i stany zagrożenia życia.
Histamina stosowana jest w celach diagnostycznych, jako dodatnia próba kontrolna podczas baań chorób alergicznych. Jest ona również stosowana w maściach jako środek powodujący zaczerwienienie i rozgrzanie skóry.
Dużo szersze zastosowanie mają leki przeciwhistaminowe, które wypierają histaminę z połączeń z receptorami. Leki antyhistaminowe pomagają w zapobieganiu i leczeniu objawów alergii i pospolitego przeziębienia. Mogą one również użyte być w leczeniu lęku i bezsenności. Stanowią one odwracalne i konkurencyjne blokery receptora H1. Nie wpływają ponadto na receptor H2. Wyróżnia się wśród nich:

Ponieważ ludzki organizm potrafi syntezować glicynę jest ona nazywana aminokwasem endogennym. Może być ona produkowana: z glioksalanu i glutaminianu przez aminotransferazę glutaminianową; z alaniny przez aminotransferazę alaninową. Ważnym sposobem syntezy glicyny u ssaków jest również synteza z choliny oraz seryny. Poniżej przedstawiony jest uproszczony szlak syntezy glicyny z choliny. Cholina ulega przemianie w betainę. Betaina ulega przemianie w sarkozynę. Ostatecznie przemiany choliny doprowadzają do powstania glicyny.
Glicyna ulega degradacji w organizmie na drodze trzech szlaków metabolicznych. Glicyna może np. ulec przekształceniu w serynę. Reakcję tę katalizuje hydroksymetylotransferaza serynowa, zaś koenzymami reakcji są tetrahydrofolian i fosforan pirydoksalu. Degradacja glicyny może jednakże również zachodzić w wątrobie za pomocą mitochondrialnego kompleksu syntazy glicynowej, który potrafi rozłożyć ją na dwutlenek węgla oraz jon amonowy, przy okazji tworząc N5,N10-metylenotetrahydrofolian. Kofaktorem tego procesu jest fosforan pirydoksalu. Owa ścieżka metaboliczna ma olbrzymie znaczenie dla ssaków, zaś jej zaburzenia są w stanie doprowadzić do hiperglicynemii. Owa ścieżka metaboliczna degradacji glicyny wraz z przedstawieniem kolorami losów poszczególnych grup atomów przedstawiona została poniżej:

Trzecim sposobem degradacji glicyny jest utlenienie przez oksydazę D-aminokwasów. Glicyna zostaje przekształcona do glioksalanu, który z kolei jest utleniany w NAD+-zależnej reakcji do szczawianu. Ponadto glicyna ulega licznym przemianom w inne metabolity, co zostało opisane w podrozdziale dotyczącym jej funkcji.
Chorobami związanymi z przemianami glicyny są schorzenia takie jak, np. Glicynuria, która wynika z zaburzeń reabsorpcji glicyny w kanalikach nerkowych i polega na wydalaniu większych aniżeli normalnie ilości tego aminokwasu. Innym zaburzeniem jest pierwotna huperoksaluria - zaburzenie katabolizmu glioksalanu, który powstaje przez deaminację glicyny. Następujące w nim utlenienie glioksalanu do szczawianu skutkuje kamicą moczową a także wapnicą nerek. Może to doprowadzić do przedwczesnych zgonów na skutek niewydolności nerek a także nadciśnienia. Inną chorobą związaną z przemianami glicyny jest nieketonowa hiperglicynemia - choroba genetyczna, która powoduje nagromadzanie się glicyny we krwi i płynie mózgowo - rdzeniowym.
Histamina (β-imidazolyloetyloamina) - jest to organiczny związek chemiczny, heterocykliczna amina, która stanowi pochodną imidazolu. Zawiera ona boczny łańcuch etyloaminowy. Występuje naturalnie w organizmie ludzkim, gdzie pełni funkcję mediatora procesów zapalnych, mediatora odczynu alergicznego, neuroprzekaźnika, a także pobudza wydzielanie kwasu żołądkowego. Histamina jest hormonem tkankowym (zwierzęcym hormonem uczestniczącym w lokalnym pozanerwowym sterowaniu działaniem narządów) zaliczanym do neurohormonów, wytwarzanym z aminokwasu histydyny. W ludzkim organizmie powstaje w wyniku dekarboksylacji (w obecności fosforanu pirydoksalu) histydyny. Jej produkcja zachodzi w wielu miejscach. Najwyższe stężenia zaobserwować można w płucach, skórze, błonie śluzowej nosa i żołądka. Histamina magazynowana jest w formie nieczynnej w ziarnistościach bazofili - granulocytów zasadochłonnych i mastocytów - komórek tucznych, które znajdują się w tkance łącznej, przede wszystkim w okolicy naczyń krwionośnych i limfatycznych, a także nerwów, skąd może uwalniana być w reakcji zapalnej. W żołądku histamina występuje w histaminocytach, zaś w ośrodkowym układzie nerwowym w neuronach histaminergicznych.
Aminy biogenne (w tym histamina) występują w wielu artykułach żywnościowych, przede wszystkim wytwarzanych i dojrzewających przy udziale procesów fermentacyjnych, a także nieświeżych, lub silnie skażonych mikrobiologicznie. Prekursorami tychże amin są aminokwasy, które uwalniane są z białek na drodze hydrolizy.
Histamina głównie występuje w surowcach rybnych. Spożycie dużej jej ilości jest powiązane z zatruciem pokarmowym. Zawartość histaminy w surowcach i przetworach rybnych zależy przede wszystkim od ilości wolnej histydyny w mięśniach, obecności aktywatorów i inhibitorów dekarboksylaz, a także od rodzajów i wielkości populacji bakteryjnej.
W wyniku reakcji antygenu z przeciwciałami we krwi człowieka lub pod wpływem czynników niszczących błonę komórki tucznej magazynującej histaminę, np wskutek zimna lub ucisku, a także pod wpływem zmian pH, składu jonów oraz leków pobudzających aktywność histaminy) dochodzi do uwalniania histaminy i rozwoju reakcji zapalnej. Histamina wyzwolona z tkanek wiąże się ze swoistymi receptorami, a następnie ulega przemianie do nieczynnych produktów. Po degranulacji komórek tucznych histamina przenika do naczyń krwionośnych, a jej poziom we krwi rośnie między 2,5 a 5 minutami i wraca do poziomu wyjściowego po 15-30 minutach. Działanie histaminy opiera się na pobudzaniu receptorów H1, H2, H3 i H4. Efekty owego pobudzenia są następujące dla poszczególnych receptorów:
Histamina uczestniczy w zapaleniu alergicznym jako mediator prozapalny nie tylko wczesnej, ale i późnej fazy odczynu, ale prawdopodobnie wpływa również na odległe następstwa choroby w postaci przebudowy dróg oddechowych. W reakcji alergicznej natychmiastowej centralną rolę odgrywa proces aktywacji receptora H1. Kontakt z alergenem powoduje wylew histaminy z ziarnistości niektórych ludzkich komórek odpornościowych, np. mastocytów i silne podrażnienie błony śluzowej jelit, nosa, oskrzeli, płuc oraz tkanki skórnej. Odpowiednio do tych lokalizacji wywołuje to: pokrzywkę skórną, biegunkę, katar sienny, astmę, a u niektórych wstrząs anafilaktyczny, np. wskutek ukąszenia osy lub pszczoły. Histamina odpowiada ponadto za wystąpienie objawów:
- alergii wziewnej oraz skórnej - wywołuje bowiem zaczerwienienie, lokalne ocieplenie, nabrzmienie i bolesny stan zapalny. Rozszerza małe naczynia krwionośne i zwiększa ich przepuszczalność; powoduje lokalne wysięki osocza i obrzęki (nosa, gardła, oskrzeli, a także obrzęk naczyniorychowy i obecność bąbli przy pokrzywce). Drażnienie przez histaminę zakończeń nerwów czuciowych powoduje świąd i doznania bólowe.
- astmy - jej nagły wylew grozi bowiem obrzękiem krtani, tchawicy, zaś w najostrzejszych przypadkach obrzękiem płuc, co odcina dopływ powietrza. Zmiany te powodują utrudnienia w oddychaniu, które stanowią główny objaw dychawicy oskrzelowej,
- alergii pokarmowej - jej obecność w jelitach powoduje bowiem skurcz mięśni gładkich i wzmożone wydzielanie soków trawiennych. Podrażniona błona śluzowa jelita cienkiego reaguje stanem zapalnym, który wywołuje biegunkę,
- anafilaksji - po przekroczeniu pewnego progu ilościowego histamina przedostaje się z tkanek do krążenia i może wywołać niebezpieczne reakcje ogólnoustrojowe, które objawiają się nagłym spadkiem ciśnienia krwi oraz przyspieszeniem akcji serca, co może grozić zgonem,
- przewlekłej alergii, np. całorocznego alergicznego nieżytu nosa lub przewlekła pokrzywka idiopatyczna - stale uwalniana histamina bowiem drastycznie zwiększa przepuszczalność ścian naczyń krwionośnych wskutek ciągłego drażnienia tzw. receptorów histaminowych H1. Przewlekły stan zapalny i drażniące działanie histaminy oraz innych mediatorów powodują stopniową degenerację okolicznych tkanek.
Histamina zmienia również właściwości błony komórkowej, wskutek czego do wnętrza komórki dostaje się zbyt dużo jonów wapnia i sodu. Może to wywoływać nadmierne skurcze mięśni oskrzeli i stany zagrożenia życia.
Histamina stosowana jest w celach diagnostycznych, jako dodatnia próba kontrolna podczas baań chorób alergicznych. Jest ona również stosowana w maściach jako środek powodujący zaczerwienienie i rozgrzanie skóry.
Dużo szersze zastosowanie mają leki przeciwhistaminowe, które wypierają histaminę z połączeń z receptorami. Leki antyhistaminowe pomagają w zapobieganiu i leczeniu objawów alergii i pospolitego przeziębienia. Mogą one również użyte być w leczeniu lęku i bezsenności. Stanowią one odwracalne i konkurencyjne blokery receptora H1. Nie wpływają ponadto na receptor H2. Wyróżnia się wśród nich:
Wyjaśnienia terminów:
1) EOZYNOFILOWE ZAPALENIE PRZEŁYKU - jest to przewlekła choroba zapalna przełyku, która charakteryzuje się występowaniem izolowanych nacieków eozynofilowych w obrębie nabłonka przełyku, a także zmian makroskopowych (błon, pierścieni), które wywołują zwężenie światła przełyku oraz objawy kliniczne choroby.
2) SEDACJA (Uspokojenie) - jest to obniżenie aktywności ośrodkowego układu nerwowego (OUN) za pomocą środków farmakologicznych bez wyłączenia świadomości (jest jednakże możliwe jej ograniczenie). Dochodzi wówczas do zmniejszenia napięcia i niepokoju, co często występuje w połączeniu z sennością. Stosowane niegdyś w tym celu leki uspokajające (sedativa), np. barbiturany, straciły obecnie na znaczeniu na rzecz małych dawek anksjolityków działających bardziej specyficznie, np. benzodiazepiny. Osobną grupę, która nadal jest w powszechnym użyciu, stanowią roślinne leki uspokajające, otrzymywane z takich roślin jak kozłek lekarski (Valeriana officinalis) - waleriana, a także chmiel zwyczajny (Humulus lupulus) czy miłek wiosenny (Adonis vernalis).
3) BILASTYNA - Bilastyna znosi wszystkie działania biologiczne histaminy. Pobudzając receptor H1, histamina powoduje skurcz mięśni gładkich oskrzeli - napad duszności, a także jelit u alergików pokarmowych. Doprowadza także do rozszerzenia naczyń tętniczych i naczyń żylnych błon śluzowych, co odpowiada za powstanie wodnistej wydzieliny z nosa i bąbli pokrzywkowych z rumieniem. Pobudzenie wybranych receptorów czuciowych skutkuje zaś świądem nosa, skóry i odruchowym kichaniem. Bilastyna blokuje biologiczne działanie histaminy i stabilizuje receptor H1 w formie nieaktywnej i uniemożliwia połączenie się z nim cząsteczki histaminy, a w konsekwencji aktywację receptora i dalsze przekazywanie sygnału.
Bilastyna znosi wszystkie działania biologiczne histaminy – jednego z najważniejszych mediatorów w alergicznym nieżycie nosa, alergicznym zapaleniu spojówek czy w astmie oskrzelowej, a także w innych procesach zapalnych. Pobudzając receptor H1, histamina powoduje skurcz mięśni gładkich oskrzeli – napad duszności, a także jelit u alergików pokarmowych. Doprowadza także do rozszerzenia naczyń tętniczych i naczyń żylnych błon śluzowych, co odpowiada za powstanie wodnistej wydzieliny z nosa i bąbli pokrzywkowych z rumieniem. Pobudzenie wybranych receptorów czuciowych skutkuje zaś świądem nosa, skóry oraz odruchowe kichanie. Bilastyna blokuje biologiczne działanie histaminy, stabilizuje receptor H1 w formie nieaktywnej, uniemożliwia połączenie się z nim cząsteczki histaminy, a w konsekwencji aktywację receptora i dalsze przekazywanie sygnału. Prowadzi to do:
- zmniejszenia obrzęku tkanek;
- zmniejszenia nacieku zapalnego;
- zmniejszenia wydzielania gruczołowego;
- zaniku kichania;
- zmniejszenia świądu skóry i błon śluzowych;
- zaniku rumienia;
- zmniejszenia obrzęku skóry, błony śluzowej nosa i tkanek objętych zapaleniem;
- obniżenia aktywności limfocytów B.
Bilastyna jako lek przeciwhistaminowy II generacji wykazuje wysokie powinowactwo do receptora histaminowego H1 i jedynie śladowe powinowactwo do receptorów innych amin i peptydów. Powinowactwo do receptora H1 jest około trzy razy większe niż cetyryzyny (antagonisty receptora H1, pochodnej hydroksyzyny; leku przeciwhistaminowego II generacji) i pięć razy większe niż feksofenadyny. Przekłada się to na obniżenie ryzyka działań niepożądanych w stosunku do leków przeciwhistaminowych I generacji, które odznaczają się niską selektywnością i działają nie tylko na receptor H1, lecz także na receptory muskarynowe, receptory adrenergiczne alfa, receptory serotoninowe i kanały potasowe. W odróżnieniu od nich bilastyna nie przenika przez barierę krew-mózg i nie blokuje receptora H1 w ośrodkowym układzie nerwowym. Wykazuje także działanie zarówno receptorowe, jak i pozareceptorowe, odznaczając się dodatkowym działaniem przeciwzapalnym, niezależnym od receptora H1 i innych receptorów histaminowych.
Istnieje możliwość zatrucia się histaminą. Histamina zawarta w żywności nie ulega bowiem rozkładowi w procesie obróbki termicznej. Spożyta wraz z pokarmem jest ona w dużym stopniu wiązana i dezaktywowana przez diaminooksydazę w przewodzie pokarmowym, co obniża jej toksyczność. W przypadku niewystarczającej aktywności diaminooksydazy, spowodowanej na przykład predyspozycjami genetycznymi, zażywaniem leków lub spożytym alkoholem, histamina może powodować efekty toksyczne. Zatrucie spowodowane histaminą wywołuje w dużych ilościach gwałtowne rozszerzenie naczyń krwionośnych i spadek ciśnienia krwi. W ilości od 200 do 1000 ppm powoduje wymioty, bóle głowy, mdłości, wysoką gorączkę, wysypki skórne, nadmierne pocenie się, trudności w oddychaniu. W przypadku obecności histaminy w mięsie w ilości powyżej 1000 ppm wywołane zatrucia pokarmowe mogą zakończyć się nawet śmiercią. Toksyczność jej bowiem zależy nie tylko od dawki, lecz również od indywidualnych predyspozycji organizmu, to znaczy funkcjonowania mechanizmu zatrzymywania i detoksykacji oraz od obecności w pokarmie innych biologicznie czynnych amin.
Hormon adrenokortykotropowy (kortykotropina - ACTH) - jest to hormon peptydowy przysadki mózgowej, który pobudza korę nadnerczy do wydzielania kortyzolu i wielu słabo działających androgenów. ACTH zbudowany jest z tylko jednego łańcucha polipeptydowego (stanowi zatem hormon peptydowy), który zawiera 39 aminokwasów w sekwencji:
- NH2-Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Try-Gly-Lys-Pro-Val-Gly-Lys-Lys-Arg-Arg-Pro-Val-Lys-Val-Tyr-Pro-Asp-Ala-Gly-Glu-Asp-Glu-Ser-Ala-Glu-Ala-Phe-Pro-Leu-Glu-Phe-COOH
Aktywność biologiczna zależna jest od 20 N-końcowych aminokwasów ACTH, które są identyczne u wszystkich gatunków (i na tej zasadzie syntetyczne analogi ACTH stosowane są w lecznictwie), zaś centrum aktywne ma 5-10 aminokwasów. ACTH jest syntetyzowany przez komórki przedniego płata przysadki mózgowej w odpowiedzi na wzrost poziomu CRH (kortykoliberyny - wielopeptydowego neuroprzekaźnika i hormonu związanego z odpowiedzią organizmu na stres) we krwi - jest to bowiem najważniejszy czynnik pobudzający uwalnianie ACTH. Hamowanie wydzielania ACTH i CRH następuje pod wpływem wzrostu we krwi stężenia ACTH, kortyzolu i innych kortykosteroidów, włączając w to steroidy egzogenne, na zasadzie sprzężenia zwrotnego. Oś CRH-ACTH-kortyzol odgrywa podstawową rolę w odpowiedzi ustroju na stres. Kortyzol m.in. podnosząc stężenie glukozy we krwi, pozwala ustrojowi przetrwać sytuację zagrożenia homeostazy. Wydzielanie ACTH odbywa się w rytmie dobowym. Skutkiem tego rytmu jest uzyskanie najwyższego stężenia kortyzolu we krwi wczesnym rankiem, kiedy organizm jest przygotowany do stresu jakim jest dla niego pobudka. Przy braku ACTH kora nadnerczy zanika i wydzielanie kortyzolu ustaje. W odpowiedzi na niewydolność kory nadnerczy lub w efekcie nadczynności przysadki następuje nadmierna synteza ACTH, która pośrednio powoduje ciemniejsze zabarwienie skóry. Jest to spowodowane tym, że ACTH syntetyzowane jest razem z MSH - hormonem tropowym, który pobudza brązowienie skóry. Jest to patogeneza jednego z objawów choroby Addisona - schorzenia układu dokrewnego, który stanowi zespół objawów spowodowanych przewlekłym niedoborem hormonów produkowanych przez korę nadnerczy, podczas którego pojawia się narastające osłabienie, skłonność do zasłabnięć (spowodowana hipotonią ortostatyczną lub hipoglikemią); osłabienie siły mięśniowej, narastająca męczliwość; utrata apetytu, spadek masy ciała, nudności, wymioty, drażliwość, biegunka, obniżenie nastroju; niedociśnienie tętnicze; przebarwienia skóry zwane jako melasma supranernale, co szczególnie dotyczy okolic eksponowanych na słońce, a także łokci, linii zgięciowych dłoni, otoczek sutkowych oraz blizn (od właśnie owych przebarwień pochodzi inna nazwa tejże choroby - cisawice; cisawy, czyli mający barwę drewna cisu); hipoglikemia (szczególnie u dzieci); zaburzenia miesiączkowania; cierpnięcie kończyn; eozynofilia, itd.
Hormon antydiuretyczny (ADH, AVP, wazopresyna, adiuretyna) - jest to organiczny związek chemiczny z grupy oligopeptydów, który u ssaków (a także u człowieka) pełni funkcję hormonu. Wytwarzany jest on przez podwzgórze w postaci preoprowazopresynoneurofizyny i uwalniany w ostatecznej postaci przez tylny płat przysadki mózgowej. Powoduje on zagęszczenie moczu pobudzając resorpcję wody w kanalikach nerkowych (kanalikach zbiorczych) poprzez pobudzanie receptorów V2. Na akwaporyny działa w sposób dwojaki, tzn. krótkoterminowa regulacja kieruje AQP2 do błony (odpowiedź następuje w ciągu kilku minut); natomiast długoterminowa (gdzie efekty widoczne są dopiero na przestrzeni kilku dni) powoduje ekspresję genów kodujących kanał AQP2 (w przeciwieństwie do AQP1 stale obecnego w błonie) dla wody, następnie translację tegoż mRNA oraz wbudowanie tego produktu w błonę bieguna aplikalnego komórek sześciennych budujących kanaliki nerkowe. Powoduje ponadto skurcz naczyń krwionośnych poprzez receptory V1, obecne na ich powierzchni. Jest również podejrzewana o udział w regulacji zachowań społecznych u różnych gatunków zwierząt, m.in. u ludzi.
Wydzielanie wazopresyny jest pobudzane przez wzrost ciśnienia osmotycznego osocza krwi i płynu mózgowo-rdzeniowego, hipowolemię lub angiotensynę II. Jej zwiększone wydzielanie następuje również podczas snu. Spadek osmolalności osocza lub hiperwolemia hamują jej wydzielanie.
Niedobór hormonu antydiuretycznego lub brak jego działania powoduje moczówkę prostą. Jeżeli dotyczy ona zaburzenia wydzielania na poziomie podwzgórza lub przysadki jest to moczówka prosta ośrodkowa. Jeżeli natomiast występuje niewrażliwość cewek nerkowych na działanie hormonu antydiuretycznego - jest to moczówka prosta nerkowa. Objawem moczówki prostej jest poliura i polidypsja. Poliura zwana wielomoczem to wydalanie moczu w ilości powyżej 3 litrów na dobę (fizjologicznie występuje przy nadmiernym spożywaniu płynów, kofeiny, alkoholu, w ciąży oraz w stanach chorobowych takich jak właśnie moczówka prosta, ale również cukrzyca i ostra niewydolność nerek). Współwystępujący często z poliurą objaw zwany polidypsją polega zaś na nadmiernym spożywaniu płynów powodowanym nadmiernym pragnieniem. Dla moczówki prostej jest również charakterystyczna niezdolność do zagęszczania moczu. Obecnie uważa się, że w 30% przyczyny moczówki prostej są nieznane, w 25% przypadków pojawia się ona w wyniku łagodnych i złośliwych guzów mózgu i/lub przysadki mózgowej, w 20% zaś po operacjach na OUN i w 16% po urazach głowy. Podstawą rozpoznania jest odpowiedź na podaną wazopresynę - obserwujemy wówczas spadek objętości moczu i wzrost jego osmolarności w moczówce centralnej, a bez wystąpienia zmian w moczówce nerkowej. Poziom wazopresyny w moczówce centralnej jest niski w stosunku do osmolarności osocza, zaś w moczówce nerkowej jest wysoki, co związane jest z utratą zdolności do zatrzymania wody z powodu utraty kanałów wodnych zależnych od wazopresyny (akwaporynach) zlokalizowanych w pęcherzykach tuż pod powierzchnią nabłonka kanalików zbiorczych.
Nadmiar wazopresyny wywołuje zespół Schwartza-Barttera, zwany również Zespołem Niewłaściwego Uwalniania Wazopresyny), który jest wywołany nadmiernym uwalnianiem hormonu antydiuretycznego (ADH) (wazopresyny, AVP) przez tylny płat przysadki mózgowej lub wydzielaniem ektopowym, hiponatremią (dosłownie - niedobór sodu we krwi) i hipoosmolarnością płynów ustrojowych. Przyczynami mogą być uszkodzenia mózgu, np. urazy, guzy, operacje, zapalenia, wodogłowie, itd.; choroby płuc - różnego rodzaju infekcje (zapalenia płuc, gruźlica, ropień), astma, ostra niewydolność oddechowa, sztuczna wentylacja; nowotwory - np. rak płuca, przewodu pokarmowego, gruczołu krokowego (prostaty), rakowiaki, chłoniaki, grasiczak; inna przyczyna: prawokomorowa niewydolność serca; kolejną przyczyną może być zażywanie różnego rodzaju leków, np. wazopresyny, chlorpropamidu, klofibratu, karbamazepiny, indapamidu, morfiny.
Katecholaminy - są to organiczne związki chemiczne stanowiące pochodne aminokwasu - tyrozyny. Niektóre z nich są aminami biogennymi. Katecholaminy są rozpuszczalne w wodzie i w 50% krążą we krwi związane z białkami osocza. W największym stężeniu występują adrenalina, noradrenalina i dopamina, które produkowane są głównie przez rdzeń nadnerczy i pozazwojowe włókna układu sympatycznego. Adrenalina działa jak neurotransmiter w centralnym systemie nerwowym, a także jako hormon regulujący krążenie krwi. Noradrenalina jest głównie neurotransmiterem w obwodowej części układu sympatycznego, ale występuje również w krwi, dzięki zjawisku wyciekania z synaps. Ważną katecholaminą jest również izoprenalina - syntetyczna pochodna adrenaliny, która nie występuje w organizmie. Wysoki poziom katecholamin we krwi wiąże się ze stresem, który może być wywołany jako reakcja psychologiczna, bądź może powstać w odpowiedzi na stresory środowiskowe, takie jak hałas czy intensywne światło. Katecholaminy wywołują ogólne reakcje organizmu, które przygotowują ciało do wysiłku fizycznego związanego z walką lub ucieczką. Typowe efekty działania to: podniesienie ciśnienia krwi, przyspieszenie akcji serca, podniesienie poziomu glukozy we krwi. Niektóre leki, np. centralne inhibitory COMT, podnoszą poziom katecholamin. Katecholaminy posiadają ponadto strukturę pierścienia benzenowego z dwiema grupami hydroksylowymi i bocznym łańcuchem etylowym połączonym z końcową grupą aminową. Okres biologicznego półtrwania katecholamin znajdujących się we krwi wynosi kilka minut.
Katecholaminy wytwarzane są z L-tyrozyny. Głównym jej źródłem jest białko pokarmowe a także fenyloalanina (ulegająca hydroksylacji w wątrobie). Po wprowadzeniu grupy wodorotlenowej do pozycji orto pierścienia benzenowego powstaje 1-3,4-dihydroksyfenyloalanina (DOPA). Reakcja ta przebiega w mitochondriach przy udziale hydroksylazy tyrozynowej, 6-metylo-tetrahydropterydyny, NADPH, tlenu cząsteczkowego i aktywatora w postaci jonów dwuwartościowego żelaza. DOPA ulega dekarboksylacji w cytoplazmie do dopaminy (1-dihydroksyfenyloetyloamina). Reakcja zachodzi przy udziale DOPA-dekarboksylazy (dekarboksylaza aromatycznych 1-aminokwasów) czy fosforanu pirydoksalu.
Dopamina jest pobierana do ziarnistości komórkowych, gdzie jej łańcuch boczny ulega utlenieniu dzięki β–hydroksylazie do 1-noradrenaliny. Reakcja zachodzi przy udziale jonów miedzi, tlenu cząsteczkowego, kwasu askorbinowego i kwasu fumarowego. Noradrenalina w cytoplazmie komórek chromochłonnych rdzenia nadnerczy, narządu Zuckerkandla i niewielkiej ilości komórek w sercu i mózgu ulega przekształceniu do adrenaliny. Proces polega na przeniesieniu grup metylowej z S-adenozynometioniny na grupę aminową noradrenaliny, przy udziale N-metylotransferazy.
Katecholaminy regulują syntezę na zasadzie ujemnego sprzężenia zwrotnego, poprzez wpływ na hydroksylazę tyrozynową i beta-hydroksylazę dopaminy. Wpływ na syntezę również mają ACTH, hormony kory nadnerczy, prostaglandyny E, angiotensyna II i bradykinina.

Katecholaminy magazynowane są w pęcherzykach ziarnistych znajdujących się w dystalnych częściach neuronów. Mogę one wychwytywać krążącą dopaminę, noradrenalinę i adrenalinę, a także, dzięki obecności beta-hydroksylazy, produkować noradrenalinę. W ziarnistościach zgromadzonych jest 2/3 ogólnej zawartości katecholamin; pozostała część znajduje się w cytoplazmie. Aminy katecholowe w ziarnistościach są wiązane z ATP, RNA i chromatograniną A. Owe połączenia chronią katecholaminy przed rozkładem.
Wyróżnia się 2 typy pęcherzyków ziarnistych. W mniejszych pęcherzykach magazynowane są wolne katecholaminy, szybko uwalniane pod wpływem pobudzenia nerwowego. W większych pęcherzykach gromadzona jest większa część amin; jest to tzw. pula magazynująca.
W rdzeniu nadnerczy magazynowana jest głównie adrenalina, podczas gdy w pozazwojowych zakończeniach nerwowych - noradrenalina.
Tlenek azotu - nieorganiczny związek chemiczny z grupy tlenków azotu, w którym azot występuje na II stopniu utlenienia. Tlenek azotu posiada ogromne znaczenie biologiczne Jest on bowiem związkiem o dużej biologicznej aktywności (ma duży wpływ na organizm). Spełnia wiele istotnych funkcji fizjologicznych u ssaków (a także u człowieka). Ze względu na niewielkie rozmiary cząsteczki i lipofilowość, tlenek azotu łatwo przenika przez błony biologiczne bez pośrednictwa układów transportujących.
W organizmie telenk azotu powstaje z grupy guanidynowej z L-argininy w reakcji katalizowanej przez kilka izoform enzymu syntazy tlenku azotu (NOS). W reakcji konieczna jest obecność tlenu cząsteczkowego oraz kofaktorów: NADPH, FAD, tetrahydrobiopteryny (BH4).

Hormon antydiuretyczny (ADH, AVP, wazopresyna, adiuretyna) - jest to organiczny związek chemiczny z grupy oligopeptydów, który u ssaków (a także u człowieka) pełni funkcję hormonu. Wytwarzany jest on przez podwzgórze w postaci preoprowazopresynoneurofizyny i uwalniany w ostatecznej postaci przez tylny płat przysadki mózgowej. Powoduje on zagęszczenie moczu pobudzając resorpcję wody w kanalikach nerkowych (kanalikach zbiorczych) poprzez pobudzanie receptorów V2. Na akwaporyny działa w sposób dwojaki, tzn. krótkoterminowa regulacja kieruje AQP2 do błony (odpowiedź następuje w ciągu kilku minut); natomiast długoterminowa (gdzie efekty widoczne są dopiero na przestrzeni kilku dni) powoduje ekspresję genów kodujących kanał AQP2 (w przeciwieństwie do AQP1 stale obecnego w błonie) dla wody, następnie translację tegoż mRNA oraz wbudowanie tego produktu w błonę bieguna aplikalnego komórek sześciennych budujących kanaliki nerkowe. Powoduje ponadto skurcz naczyń krwionośnych poprzez receptory V1, obecne na ich powierzchni. Jest również podejrzewana o udział w regulacji zachowań społecznych u różnych gatunków zwierząt, m.in. u ludzi.
Wydzielanie wazopresyny jest pobudzane przez wzrost ciśnienia osmotycznego osocza krwi i płynu mózgowo-rdzeniowego, hipowolemię lub angiotensynę II. Jej zwiększone wydzielanie następuje również podczas snu. Spadek osmolalności osocza lub hiperwolemia hamują jej wydzielanie.
Niedobór hormonu antydiuretycznego lub brak jego działania powoduje moczówkę prostą. Jeżeli dotyczy ona zaburzenia wydzielania na poziomie podwzgórza lub przysadki jest to moczówka prosta ośrodkowa. Jeżeli natomiast występuje niewrażliwość cewek nerkowych na działanie hormonu antydiuretycznego - jest to moczówka prosta nerkowa. Objawem moczówki prostej jest poliura i polidypsja. Poliura zwana wielomoczem to wydalanie moczu w ilości powyżej 3 litrów na dobę (fizjologicznie występuje przy nadmiernym spożywaniu płynów, kofeiny, alkoholu, w ciąży oraz w stanach chorobowych takich jak właśnie moczówka prosta, ale również cukrzyca i ostra niewydolność nerek). Współwystępujący często z poliurą objaw zwany polidypsją polega zaś na nadmiernym spożywaniu płynów powodowanym nadmiernym pragnieniem. Dla moczówki prostej jest również charakterystyczna niezdolność do zagęszczania moczu. Obecnie uważa się, że w 30% przyczyny moczówki prostej są nieznane, w 25% przypadków pojawia się ona w wyniku łagodnych i złośliwych guzów mózgu i/lub przysadki mózgowej, w 20% zaś po operacjach na OUN i w 16% po urazach głowy. Podstawą rozpoznania jest odpowiedź na podaną wazopresynę - obserwujemy wówczas spadek objętości moczu i wzrost jego osmolarności w moczówce centralnej, a bez wystąpienia zmian w moczówce nerkowej. Poziom wazopresyny w moczówce centralnej jest niski w stosunku do osmolarności osocza, zaś w moczówce nerkowej jest wysoki, co związane jest z utratą zdolności do zatrzymania wody z powodu utraty kanałów wodnych zależnych od wazopresyny (akwaporynach) zlokalizowanych w pęcherzykach tuż pod powierzchnią nabłonka kanalików zbiorczych.
Nadmiar wazopresyny wywołuje zespół Schwartza-Barttera, zwany również Zespołem Niewłaściwego Uwalniania Wazopresyny), który jest wywołany nadmiernym uwalnianiem hormonu antydiuretycznego (ADH) (wazopresyny, AVP) przez tylny płat przysadki mózgowej lub wydzielaniem ektopowym, hiponatremią (dosłownie - niedobór sodu we krwi) i hipoosmolarnością płynów ustrojowych. Przyczynami mogą być uszkodzenia mózgu, np. urazy, guzy, operacje, zapalenia, wodogłowie, itd.; choroby płuc - różnego rodzaju infekcje (zapalenia płuc, gruźlica, ropień), astma, ostra niewydolność oddechowa, sztuczna wentylacja; nowotwory - np. rak płuca, przewodu pokarmowego, gruczołu krokowego (prostaty), rakowiaki, chłoniaki, grasiczak; inna przyczyna: prawokomorowa niewydolność serca; kolejną przyczyną może być zażywanie różnego rodzaju leków, np. wazopresyny, chlorpropamidu, klofibratu, karbamazepiny, indapamidu, morfiny.
Katecholaminy - są to organiczne związki chemiczne stanowiące pochodne aminokwasu - tyrozyny. Niektóre z nich są aminami biogennymi. Katecholaminy są rozpuszczalne w wodzie i w 50% krążą we krwi związane z białkami osocza. W największym stężeniu występują adrenalina, noradrenalina i dopamina, które produkowane są głównie przez rdzeń nadnerczy i pozazwojowe włókna układu sympatycznego. Adrenalina działa jak neurotransmiter w centralnym systemie nerwowym, a także jako hormon regulujący krążenie krwi. Noradrenalina jest głównie neurotransmiterem w obwodowej części układu sympatycznego, ale występuje również w krwi, dzięki zjawisku wyciekania z synaps. Ważną katecholaminą jest również izoprenalina - syntetyczna pochodna adrenaliny, która nie występuje w organizmie. Wysoki poziom katecholamin we krwi wiąże się ze stresem, który może być wywołany jako reakcja psychologiczna, bądź może powstać w odpowiedzi na stresory środowiskowe, takie jak hałas czy intensywne światło. Katecholaminy wywołują ogólne reakcje organizmu, które przygotowują ciało do wysiłku fizycznego związanego z walką lub ucieczką. Typowe efekty działania to: podniesienie ciśnienia krwi, przyspieszenie akcji serca, podniesienie poziomu glukozy we krwi. Niektóre leki, np. centralne inhibitory COMT, podnoszą poziom katecholamin. Katecholaminy posiadają ponadto strukturę pierścienia benzenowego z dwiema grupami hydroksylowymi i bocznym łańcuchem etylowym połączonym z końcową grupą aminową. Okres biologicznego półtrwania katecholamin znajdujących się we krwi wynosi kilka minut.
Katecholaminy wytwarzane są z L-tyrozyny. Głównym jej źródłem jest białko pokarmowe a także fenyloalanina (ulegająca hydroksylacji w wątrobie). Po wprowadzeniu grupy wodorotlenowej do pozycji orto pierścienia benzenowego powstaje 1-3,4-dihydroksyfenyloalanina (DOPA). Reakcja ta przebiega w mitochondriach przy udziale hydroksylazy tyrozynowej, 6-metylo-tetrahydropterydyny, NADPH, tlenu cząsteczkowego i aktywatora w postaci jonów dwuwartościowego żelaza. DOPA ulega dekarboksylacji w cytoplazmie do dopaminy (1-dihydroksyfenyloetyloamina). Reakcja zachodzi przy udziale DOPA-dekarboksylazy (dekarboksylaza aromatycznych 1-aminokwasów) czy fosforanu pirydoksalu.
Dopamina jest pobierana do ziarnistości komórkowych, gdzie jej łańcuch boczny ulega utlenieniu dzięki β–hydroksylazie do 1-noradrenaliny. Reakcja zachodzi przy udziale jonów miedzi, tlenu cząsteczkowego, kwasu askorbinowego i kwasu fumarowego. Noradrenalina w cytoplazmie komórek chromochłonnych rdzenia nadnerczy, narządu Zuckerkandla i niewielkiej ilości komórek w sercu i mózgu ulega przekształceniu do adrenaliny. Proces polega na przeniesieniu grup metylowej z S-adenozynometioniny na grupę aminową noradrenaliny, przy udziale N-metylotransferazy.
Katecholaminy regulują syntezę na zasadzie ujemnego sprzężenia zwrotnego, poprzez wpływ na hydroksylazę tyrozynową i beta-hydroksylazę dopaminy. Wpływ na syntezę również mają ACTH, hormony kory nadnerczy, prostaglandyny E, angiotensyna II i bradykinina.

Katecholaminy magazynowane są w pęcherzykach ziarnistych znajdujących się w dystalnych częściach neuronów. Mogę one wychwytywać krążącą dopaminę, noradrenalinę i adrenalinę, a także, dzięki obecności beta-hydroksylazy, produkować noradrenalinę. W ziarnistościach zgromadzonych jest 2/3 ogólnej zawartości katecholamin; pozostała część znajduje się w cytoplazmie. Aminy katecholowe w ziarnistościach są wiązane z ATP, RNA i chromatograniną A. Owe połączenia chronią katecholaminy przed rozkładem.
Wyróżnia się 2 typy pęcherzyków ziarnistych. W mniejszych pęcherzykach magazynowane są wolne katecholaminy, szybko uwalniane pod wpływem pobudzenia nerwowego. W większych pęcherzykach gromadzona jest większa część amin; jest to tzw. pula magazynująca.
W rdzeniu nadnerczy magazynowana jest głównie adrenalina, podczas gdy w pozazwojowych zakończeniach nerwowych - noradrenalina.
Tlenek azotu - nieorganiczny związek chemiczny z grupy tlenków azotu, w którym azot występuje na II stopniu utlenienia. Tlenek azotu posiada ogromne znaczenie biologiczne Jest on bowiem związkiem o dużej biologicznej aktywności (ma duży wpływ na organizm). Spełnia wiele istotnych funkcji fizjologicznych u ssaków (a także u człowieka). Ze względu na niewielkie rozmiary cząsteczki i lipofilowość, tlenek azotu łatwo przenika przez błony biologiczne bez pośrednictwa układów transportujących.
W organizmie telenk azotu powstaje z grupy guanidynowej z L-argininy w reakcji katalizowanej przez kilka izoform enzymu syntazy tlenku azotu (NOS). W reakcji konieczna jest obecność tlenu cząsteczkowego oraz kofaktorów: NADPH, FAD, tetrahydrobiopteryny (BH4).
Zanim odkryto, że tlenek azotu jest produkowany w organizmie, istniało określenie na nieznany związek powodujący rozkurcz mięśniówki gładkiej - śródbłonkowy czynnik rozluźniający (EDRF - Endothelium - derived relaxing factor), który później okazał się być tlenkiem azotu. EDRF odkrył i scharakteryzował Robert F. Furchgott, który wraz ze swoimi współpracownikami - Louisem Ignarro i Feridem Muradem otrzymał za to Nagrodę Nobla w 1998 roku. Louis Ignarro kontynuował prace Roberta Furchgotta nad rozszerzaniem naczyń krwionośnych i wykazał, iż czynnikiem odpowiedzialnym za ten proces jest tlenek azotu. Było to pierwsze odkrycie, które wskazywało na to, że gaz może być czynnikiem sygnałowym w organizmie. Dalsze prace nad tlenkiem azotu wskazywały przyczyny i metody leczenia chorób serca, wstrząsów psychicznych i nowotworów. Praktycznym wykorzystaniem prac Furchgotta, Ignarro i trzeciego badacza - Ferida Murada, było wynalezienie leku na impotencję - sildenafilu (Viagra).
Tlenek azotu w fizjologii i stanach chorobowych:
- reguluje napięcia naczyń krwionośnych i ciśnienie tętnicze krwi,
- hamuje agregację płytek krwi i leukocytów,
- w ośrodkowym układzie nerwowym pełni funkcję neuromodulatora (wywiera wpływ m.in. na pamięć),
- w obwodowym układzie nerwowym działa jak neurotransmiter i wpływa na motorykę przewodu pokarmowego, funkcje neuroendokrynne i lokalny przepływ krwi,
- ma wpływ na wiele mechanizmów immunologicznych,
- bierze udział w procesie erekcji członka.
Źródła:
- Ben Best - Brain Neurotransmitters: http://www.benbest.com/science/anatmind/anatmd10.html,
- - The Neurotransmitter Collection - The Florida State University - Molecular Expressions: https://micro.magnet.fsu.edu/micro/gallery/neurotrans/neurotrans.html
- - Wikipedia (PL) i (EN),
- - Neuropsychologia.org,
- - "Biochemia - Krótki kurs" - J.L. Tymoczko; J.M. Berg;l L. Stryer,
- - "Zarys biochemii" - Peter Karlson,
- - Władysław Zygmunt Traczyk, Andrzej Trzebski, Fizjologia człowieka z elementami fizjologii stosowanej i klinicznej, 2009.
- - Farmakopea Polska X, Polskie Towarzystwo Farmaceutyczne, Warszawa: Urząd Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych, 2014, s. 4276, ISBN 978-83-63724-47-4.
- - Towarzystwo Farmaceutyczne: "Farmakopea Polska IX - Warszawa: Urząd Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych, 2011, s. 4574,
- - Zaawansowane zabiegi resuscytacyjne – algorytm postępowania w tachykardii, Polska Rada Resuscytacji za European Resuscitation Council, 2010 [dostęp 2012-12-16].
- - Joseph D. Losek i inni, Adenosine and Pediatric Supraventricular Tachycardia in the Emergency Department: Multicenter Study and Review, „Annals of Emergency Medicine”, 33 (2), s. 185–191, DOI: 10.1016/s0196-0644(99)70392-6, PMID: 9922414
- - Baza PubChem, United States National Library of Medicine - opisy poszczególnych neuroprzekaźników,
- - Informacje o substancjach aktywnych - DrugBank,
- - Wielka encyklopedia zdrowia. Wojciech Twardosz (red.) T. I: Ab-Az Poznań: Wydawnictwo HORYZONT, 2002 - s. 12-13ISBN 83-89242-01-X.
- - Mała encyklopedia medycyny. Wyd. IV. T. I: A–G. Warszawa: PWN, 1988, s. 11. ISBN 83-01-08835-4.
- -Szymonowicz W, Cybulski N: O funkcji nadnercza. 1895.
- - Katedra Historii Medycyny CM UJ - Fizjologia,
- - Bolesława Arabska-Przedpełska, Halina Pawlicka: Współczesna endodoncja w praktyce. Łódź. Bestom DENTOnet.pl , 2011, s. 133. ISBN 978-83-927915-6-0.
- - Anandamid (nr A0580) (ang.) – karta charakterystyki produktu Sigma-Aldrich (Merck KGaA) na obszar Stanów Zjednoczonych (ze względu na zmianę sposobu wywołania karty charakterystyki, aby pobrać kartę dla obszaru USA, na stronie produktu należy zmienić lokalizację na "United States" i ponownie pobrać kartę). [dostęp 2012-02-13].
- - P.B. Sparling i inni, Exercise activates the endocannabinoid system, „NeuroReport”, 14 (17), 2003, s. 2209-2211 [dostęp 2017-07-25] (ang.).
- - David Terraso: Research Locates Source of Runner's High Experienced by Athletes (ang.). Georgia Institute of Technology, 2004-01-08. [dostęp 2017-07-25].
- Devane i inni, Isolation and structure of a brain constituent that binds to the cannabinoid receptor, „Science”, 258, 1992, s. 1946–1949, DOI: 10.1126/science.1470919, PMID: 1470919.
- R. Mechoulam, E. Fride: The unpaved road to the endogenous brain cannabinoid ligands, the anandamides. W: Cannabinoid Receptors. R. Pertwee (red.). London: Academic Press, 1995, s. 233–258. ISBN 978-0125514606.
- Stella, N., Schweitzer, P., Piomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nature 388 773-778 (1997).
- Sugiura, T., Kodaka, T., Nakane, S., et al. Evidence that the cannabinoid CB1 receptor is a 2-arachidonoylglycerol receptor. Structure-activity relationship of 2-arachidonoylglycerol, ether-linked analogues, and related compounds. J Biol Chem 274 2794-2801 (1999).
- Dopamina (nr H8502) – karta charakterystyki produktu Sigma-Aldrich (Merck KGaA) na obszar Polski.
- ↑ a b Dopamina (CID: 681) (ang.) w bazie PubChem, United States National Library of Medicine.
- ↑ a b c d Dopamina (DB00988) – informacje o substancji aktywnej (ang.). DrugBank.
- ↑ Dopamina (ang.) w bazie ChemIDplus, United States National Library of Medicine. [dostęp 2012-08-03].
- ↑ a b Wojciech Kostowski, Zbigniew S. Herman: Farmakologia – podstawy farmakoterapii: podręcznik dla studentów medycyny i lekarzy. Wyd. 3 poprawione i uzupełnione. Warszawa: Wydawnictwo Lekarskie PZWL, 2006, s. 1569. ISBN 83-200-3352-7.
- ↑ Dopamine (ang.). PubChem.
- ↑ Catecholamine (ang.). Brittanica.
- ↑ Phenylethylamine (ang.). ChemicalLand21.com.
- ↑ a b c d e f g Dopamine Hydrochloride. „Analytical Profiles of Drug Substances”. 11, s. 257–272, 1982 (ang.).
- ↑ a b c d e f Chapter 1: Historical overview: Introduction to the dopamine receptors. W: The Dopamine Receptors. Springer, 2009, s. 1–22. ISBN 1-60327-333-6. (ang.)
- ↑ a b c d e f g h Chapter 1: Enzymes involved in the biosynthesis and degradation of catecholamines. W: Biochemistry of Biogenic Amines. Springer, 2013, s. 1–35. ISBN 1-4684-3171-4. (ang.)
- ↑ a b c Symptomatic pharmacological therapy in Parkinson’s disease. W: Parkinson's Disease. London: Royal College of Physicians, 2006, s. 59–100. ISBN 1-86016-283-5. (ang.)
- Graeme Eisenhofer, Irwin J. Kopin, David S. Goldstein, Catecholamine metabolism: a contemporary view with implications for physiology and medicine, „Pharmacological Reviews”, 56 (3), 2004, s. 331–349, DOI: 10.1124/pr.56.3.1, PMID: 15317907 (ang.).
- F. Amin, M. Davidson, K.L. Davis, Homovanillic acid measurement in clinical research: a review of methodology, „Schizophrenia Bulletin”, 18 (1), 1992, s. 123–148, DOI: 10.1093/schbul/18.1.123, PMID: 1553492 (ang.).
- F. Amin i inni, Assessment of the Central Dopaminergic Index of Plasma HVA in Schizophrenia, „Schizophrenia Bulletin”, 21 (1), 1995, s. 53–66, DOI: 10.1093/schbul/21.1.53, PMID: 7770741 (ang.).
- Intraneuronal dopamine-quinone synthesis: a review. „Neurotoxicity Research”. 1 (3), s. 181–95, 2000. DOI: 10.1007/BF03033289. PMID: 12835101 (ang.).
- Ikuko Miyazaki, Masato Asanuma, Dopaminergic neuron-specific oxidative stress caused by dopamine itself, „Acta Medica Okayama”, 62 (3), 2008, s. 141–150, DOI: 10.18926/AMO/30942, PMID: 18596830 (ang.).
- Dopamine: Biological activity (ang.). International Union of Basic and Clinical Pharmacology.
- David K. Grandy, Gregory M. Miller, Jun-Xu Li, "TAARgeting Addiction"--The Alamo Bears Witness to Another Revolution: An Overview of the Plenary Symposium of the 2015 Behavior, Biology and Chemistry Conference, „Drug and Alcohol Dependence”, 159, 2016, s. 9–16, DOI: 10.1016/j.drugalcdep.2015.11.014, PMID: 26644139, PMCID: PMC4724540 (ang.).
- Chapter 6: Dopamine receptor signalling: intracellular pathways to behavior. W: The Dopamine Receptors. Springer, 2009, s. 137–174. ISBN 1-60327-333-6. (ang.)
- Lee E. Eiden i inni, The vesicular amine transporter family (SLC18): amine/proton antiporters required for vesicular accumulation and regulated exocytotic secretion of monoamines and acetylcholine, „Pflugers Archiv: European Journal of Physiology”, 447 (5), 2004, s. 636–640, DOI: 10.1007/s00424-003-1100-5, PMID: 12827358 (ang.).
- David K. Grandy, Gregory M. Miller, Jun-Xu Li, "TAARgeting Addiction"--The Alamo Bears Witness to Another Revolution: An Overview of the Plenary Symposium of the 2015 Behavior, Biology and Chemistry Conference, „Drug and Alcohol Dependence”, 159, 2016, s. 9–16, DOI: 10.1016/j.drugalcdep.2015.11.014, PMID: 26644139, PMCID: PMC4724540 (ang.).
- Gregory M. Miller, The emerging role of trace amine-associated receptor 1 in the functional regulation of monoamine transporters and dopaminergic activity, „Journal of Neurochemistry”, 116 (2), 2011, s. 164–176, DOI: 10.1111/j.1471-4159.2010.07109.x, PMID: 21073468, PMCID: PMC3005101 (ang.).
- Jean-Martin Beaulieu, Raul R. Gainetdinov, The physiology, signaling, and pharmacology of dopamine receptors, „Pharmacological Reviews”, 63 (1), 2011, s. 182–217, DOI: 10.1124/pr.110.002642, PMID: 21303898 (ang.).
- Gonzalo E. Torres, Raul R. Gainetdinov, Marc G. Caron, Plasma membrane monoamine transporters: structure, regulation and function, „Nature Reviews. Neuroscience”, 4 (1), 2003, s. 13–25, DOI: 10.1038/nrn1008, PMID: 12511858 (ang.).
- M.E. Rice, J.C. Patel, S.J. Cragg, Dopamine release in the basal ganglia, „Neuroscience”, 198, 2011, s. 112–137, DOI: 10.1016/j.neuroscience.2011.08.066, PMID: 21939738, PMCID: PMC3357127 (ang.).
- Wolfram Schultz, Multiple dopamine functions at different time courses, „Annual Review of Neuroscience”, 30, 2007, s. 259–288, DOI: 10.1146/annurev.neuro.28.061604.135722, PMID: 17600522 (ang.).
- Anders Björklund, Stephen B. Dunnett, Dopamine neuron systems in the brain: an update, „Trends in Neurosciences”, 30 (5), 2007, s. 194–202, DOI: 10.1016/j.tins.2007.03.006, PMID: 17408759 (ang.).
- A. Dahlström, K. Fuxe. Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of brain stem neurons. „Acta Physiologica Scandinavica”. 62 (Suppl. 232), s. 1–55, 1964. PMID: 14229500 (ang.).
- Chapter 6: Widely Projecting Systems: Monoamines, Acetylcholine, and Orexin. W: Molecular Neuropharmacology: A Foundation for Clinical Neuroscience. Wyd. 2nd. New York: McGraw-Hill Medical, 2009, s. 147–148, 154–157. ISBN 0-07-148127-3.(ang.)
- Chadwick W. Christine, Michael J. Aminoff, Clinical differentiation of parkinsonian syndromes: prognostic and therapeutic relevance, „The American Journal of Medicine”, 117 (6), 2004, s. 412–419, DOI: 10.1016/j.amjmed.2004.03.032, PMID: 15380498 (ang.).
- Dopamine and the spinal cord in restless legs syndrome: does spinal cord physiology reveal a basis for augmentation?. „Sleep Medicine Reviews”. 10 (3), s. 185–96, 2006. DOI: 10.1016/j.smrv.2006.01.004. PMID: 16762808 (ang.).
- N. Ben-Jonathan, R. Hnasko, Dopamine as a prolactin (PRL) inhibitor, „Endocrine Reviews”, 22 (6), 2001, s. 724–763, DOI: 10.1210/edrv.22.6.0451, PMID: 11739329 (ang.).
- Paul Witkovsky, Dopamine and retinal function, „Documenta Ophthalmologica. Advances in Ophthalmology”, 108 (1), 2004, s. 17–40, DOI: 10.1023/B:DOOP.0000019487.88486.0a, PMID: 15104164 (ang.).
- Basal Ganglia and the Striatal Motor System. W: Neuroanatomy (Board Review Series). Wyd. 4th. Baltimore: Wulters Kluwer & Lippincott Wiliams & Wilkins, 2008, s. 274–281. ISBN 0-7817-7245-1. (ang.)
- V.S. Chakravarthy, Denny Joseph, Raju S. Bapi, What do the basal ganglia do? A modeling perspective, „Biological Cybernetics”, 103 (3), 2010, s. 237–253, DOI: 10.1007/s00422-010-0401-y, PMID: 20644953 (ang.).
- W ten sposób jądra podstawne są odpowiedzialne za rozpoczęcie ruchu, ale nie determinują w szczegółach jak zostanie on wykonanyStan B. Floresco, The nucleus accumbens: an interface between cognition, emotion, and action, „Annual Review of Psychology”, 66, 2015, s. 25–52, DOI: 10.1146/annurev-psych-010213-115159, PMID: 25251489 (ang.).Skocz do:Bernard W Balleine i inni, Hierarchical control of goal-directed action in the cortical–basal ganglia network, „Current Opinion in Behavioral Sciences”, 5, 2015, s. 1–7, DOI: 10.1016/j.cobeha.2015.06.001 (ang.).
- Jankovic, Parkinson's disease: clinical features and diagnosis, „Journal of Neurology, Neurosurgery, and Psychiatry”, 79 (4), 2008, s. 368–376, DOI: 10.1136/jnnp.2007.131045, PMID: 18344392 (ang.).
- Tommy Pattij, Louk J.M.J. Vanderschuren, The neuropharmacology of impulsive behaviour, „Trends in Pharmacological Sciences”, 29 (4), 2008, s. 192–199, DOI: 10.1016/j.tips.2008.01.002, PMID: 18304658.
- Wolfram Schultz, Neuronal Reward and Decision Signals: From Theories to Data, „Physiological Reviews”, 95 (3), 2015, s. 853–951, DOI: 10.1152/physrev.00023.2014, PMID: 26109341, PMCID: PMC4491543, Cytat: Rewards are crucial objects that induce learning, approach behavior, choices, and emotions. Whereas emotions are difficult to investigate in animals, the learning function is mediated by neuronal reward prediction error signals which implement basic constructs of reinforcement learning theory. These signals are found in dopamine neurons, which emit a global reward signal to striatum and frontal cortex, and in specific neurons in triatum, amygdala, and frontal cortex projecting to select neuronal populations ... FIGURE 12. Reward components inducing the two phasic dopamine response components. The initial component (blue) detects the event before having identified its value. It increases with sensory impact (physical salience), novelty (novelty/surprise salience), generalization to rewarded stimuli, and reward context. This component is coded as temporal event prediction error (389). The second component (red) codes reward value (as reward prediction error) ... The salience of rewards derives from three principal factors, namely, their physical intensity and impact (physical salience), their novelty and surprise (novelty/surprise salience), and their general motivational impact shared with punishers (motivational salience). A separate form not included in this scheme, incentive salience, primarily addresses dopamine function in addiction and refers only to approach behavior (as opposed to learning) (ang.).
- T.E. Robinson, K.C. Berridge, The neural basis of drug craving: an incentive-sensitization theory of addiction, „Brain Research. Brain Research Reviews”, 18 (3), 1993, s. 247–291, DOI: 10.1016/0165-0173(93)90013-p, PMID: 8401595 (ang.).
- Jason S. Wright, Jaak Panksepp, An Evolutionary Framework to Understand Foraging, Wanting, and Desire: The Neuropsychology of the SEEKING System, „Neuropsychoanalysis”, 14 (1), 2012, s. 5–39, DOI: 10.1080/15294145.2012.10773683 (ang.).
- Molecular Neuropharmacology: A Foundation for Clinical Neuroscience. Wyd. 2nd. New York: McGraw-Hill Medical, 2009, s. 147–148, 366–367, 375–376. ISBN 978-0-07-148127-4. Cytat: VTA DA neurons play a critical role in motivation, reward-related behavior (Chapter 15), attention, and multiple forms of memory. This organization of the DA system, wide projection from a limited number of cell bodies, permits coordinated responses to potent new rewards. Thus, acting in diverse terminal fields, dopamine confers motivational salience (“wanting”) on the reward itself or associated cues (nucleus accumbens shell region), updates the value placed on different goals in light of this new experience (orbital prefrontal cortex), helps consolidate multiple forms of memory (amygdala and hippocampus), and encodes new motor programs that will facilitate obtaining this reward in the future (nucleus accumbens core region and dorsal striatum). In this example, dopamine modulates the processing of sensorimotor information in diverse neural circuits to maximize the ability of the organism to obtain future rewards. ...
The brain reward circuitry that is targeted by addictive drugs normally mediates the pleasure and strengthening of behaviors associated with natural reinforcers, such as food, water, and sexual contact. Dopamine neurons in the VTA are activated by food and water, and dopamine release in the NAc is stimulated by the presence of natural reinforcers, such as food, water, or a sexual partner. ...
The NAc and VTA are central components of the circuitry underlying reward and memory of reward. As previously mentioned, the activity of dopaminergic neurons in the VTA appears to be linked to reward prediction. The NAc is involved in learning associated with reinforcement and the modulation of motoric responses to stimuli that satisfy internal homeostatic needs. The shell of the NAc appears to be particularly important to initial drug actions within reward circuitry; addictive drugs appear to have a greater effect on dopamine release in the shell than in the core of the NAc. ... If motivational drive is described in terms of wanting, and hedonic evaluation in terms of liking, it appears that wanting can be dissociated from liking and that dopamine may influence these phenomena differently. Differences between wanting and liking are confirmed in reports by human addicts, who state that their desire for drugs (wanting) increases with continued use even when pleasure (liking) decreases because of tolerance.. (ang.) - Kent C. Berridge, Terry E. Robinson, J. Wayne Aldridge, Dissecting components of reward: 'liking', 'wanting', and learning, „Current Opinion in Pharmacology”, 9 (1), 2009, s. 65–73, DOI: 10.1016/j.coph.2008.12.014, PMID: 19162544, PMCID: PMC2756052, Cytat: Conversely, amplification of ‘wanting’ without ‘liking’ has been produced by the activation of dopamine systems by amphetamine or similar catecholamine-activating drugs given systemically or microinjected directly into the nucleus accumbens, or by genetic mutation that raises extracellular levels of dopamine (via knockdown of dopamine transporters in the synapse) in mesocorticolimbic circuits, and by the near-permanent sensitization of mesocorticolimbic-dopamine-related systems by repeated administration of high-doses of addictive drugs (Figure 3–Figure 5) [39•,40•,61•,66]. We have proposed that in susceptible individuals the neural sensitization of incentive salience by drugs of abuse may generate compulsive ‘wanting’ to take more drugs, whether or not the same drugs are correspondingly ‘liked’, and thus contribute to addiction [39•,40•,42] (Figure 5).(ang.).
- Ethan S. Bromberg-Martin, Masayuki Matsumoto, Okihide Hikosaka, Dopamine in motivational control: rewarding, aversive, and alerting, „Neuron”, 68 (5), 2010, s. 815–834, DOI: 10.1016/j.neuron.2010.11.022, PMID: 21144997, PMCID: PMC3032992(ang.).
- L.M. Yager i inni, The ins and outs of the striatum: role in drug addiction, „Neuroscience”, 301, 2015, s. 529–541, DOI: 10.1016/j.neuroscience.2015.06.033, PMID: 26116518, PMCID: PMC4523218 (ang.).
- Michael P. Saddoris i inni, Differential Dopamine Release Dynamics in the Nucleus Accumbens Core and Shell Reveal Complementary Signals for Error Prediction and Incentive Motivation, „The Journal of Neuroscience: The Official Journal of the Society for Neuroscience”, 35 (33), 2015, s. 11572–11582, DOI: 10.1523/JNEUROSCI.2344-15.2015, PMID: 26290234, PMCID: PMC4540796, Cytat: Here, we have found that real-time dopamine release within the nucleus accumbens (a primary target of midbrain dopamine neurons) strikingly varies between core and shell subregions. In the core, dopamine dynamics are consistent with learning-based theories (such as reward prediction error) whereas in the shell, dopamine is consistent with motivation-based theories (e.g., incentive salience). (ang.).
- Kent C. Berridge, Morten L. Kringelbach, Pleasure systems in the brain, „Neuron”, 86 (3), 2015, s. 646–664, DOI: 10.1016/j.neuron.2015.02.018, PMID: 25950633, PMCID: PMC4425246 (ang.).
- Roy A. Wise, Dopamine and reward: the anhedonia hypothesis 30 years on, „Neurotoxicity Research”, 14 (2-3), 2008, s. 169–183, DOI: 10.1007/BF03033808, PMID: 19073424, PMCID: PMC3155128 (ang.).
- Oscar Arias-Carrión, Ernst Pŏppel, Dopamine, learning, and reward-seeking behavior, „Acta Neurobiologiae Experimentalis”, 67 (4), 2007, s. 481–488, PMID: 18320725 (ang.).
- J.D. Salamone i inni, Nucleus accumbens dopamine and the regulation of effort in food-seeking behavior: implications for studies of natural motivation, psychiatry, and drug abuse, „The Journal of Pharmacology and Experimental Therapeutics”, 305 (1), 2003, s. 1–8, DOI: 10.1124/jpet.102.035063, PMID: 12649346 (ang.).
- Chandrani Sarkar i inni, The immunoregulatory role of dopamine: an update, „Brain, Behavior, and Immunity”, 24 (4), 2010, s. 525–528, DOI: 10.1016/j.bbi.2009.10.015, PMID: 19896530, PMCID: PMC2856781 (ang.).
- Tahir Hussain, Mustafa F. Lokhandwala, Renal dopamine receptors and hypertension, „Experimental Biology and Medicine”, 228 (2), 2003, s. 134–142, DOI: 10.1177/153537020322800202, PMID: 12563019 (ang.).
- Marcelo Roberto Choi i inni, Renal dopaminergic system: Pathophysiological implications and clinical perspectives, „World Journal of Nephrology”, 4 (2), 2015, s. 196–212, DOI: 10.5527/wjn.v4.i2.196, PMID: 25949933, PMCID: PMC4419129 (ang.).
- R.M. Carey, Theodore Cooper Lecture: Renal dopamine system: paracrine regulator of sodium homeostasis and blood pressure, „Hypertension”, 38 (3), 2001, s. 297–302, DOI: 10.1161/hy0901.096422, PMID: 11566894 (ang.).
- Blanca Rubí, Pierre Maechler, Minireview: new roles for peripheral dopamine on metabolic control and tumor growth: let's seek the balance, „Endocrinology”, 151 (12), 2010, s. 5570–5581, DOI: 10.1210/en.2010-0745, PMID: 21047943 (ang.).
- Merims D, Giladi N. Dopamine dysregulation syndrome, addiction and behavioral changes in Parkinson's disease. „Parkinsonism & Related Disorders”. 14 (4), s. 273-280, 2008. DOI: 10.1016/j.parkreldis.2007.09.007. PMID: 17988927.
- Jing Wu i inni, Role of dopamine receptors in ADHD: a systematic meta-analysis, „Molecular Neurobiology”, 45 (3), 2012, s. 605–620, DOI: 10.1007/s12035-012-8278-5, PMID: 22610946.
- Craig W. Berridge, David M. Devilbiss, Psychostimulants as cognitive enhancers: the prefrontal cortex, catecholamines, and attention-deficit/hyperactivity disorder, „Biological Psychiatry”, 69 (12), 2011, e101–111, DOI: 10.1016/j.biopsych.2010.06.023, PMID: 20875636, PMCID: PMC3012746.
- S.K. Jääskeläinen i inni, Role of the dopaminergic system in chronic pain - a fluorodopa-PET study, „Pain”, 90 (3), 2001, s. 257–260, DOI: 10.1016/S0304-3959(00)00409-7, PMID: 11207397.
- Patrick B. Wood, Role of central dopamine in pain and analgesia, „Expert Review of Neurotherapeutics”, 8 (5), 2008, s. 781–797, DOI: 10.1586/14737175.8.5.781, PMID: 18457535.
- Disruption of gene interaction linked to schizophrenia (ang.). St. Jude Children's Research Hospital. [dostęp 2006-07-06].
- J.W. Maas i inni, Schizophrenia, Psychosis, and Cerebral Spinal Fluid Homovanillic Acid Concentrations, „Schizophrenia Bulletin”, 23 (1), 1997, s. 147–154, DOI: 10.1093/schbul/23.1.147, PMID: 9050120 (ang.).
- Dopamine Receptor Blockade: Antipsychotic Drugs, www.williams.edu [dostęp 2017-11-22].
- Dopamine and antipsychotic drug action revisited | The British Journal of Psychiatry, bjp.rcpsych.org [dostęp 2017-11-22] (ang.).
- M.J. Durcan i inni, Is clozapine selective for the dopamine D4 receptor?, „Life Sciences”, 57 (18), 1995, PL275–283, DOI: 10.1016/0024-3205(95)02151-8, PMID: 7475902 (ang.).
- Methamphetamine 101
- J.A. Lieberman, J.M. Kane, J. Alvir, Provocative tests with psychostimulant drugs in schizophrenia, „Psychopharmacology”, 91 (4), 1987, s. 415–433, DOI: 10.1007/BF00216006, PMID: 2884687 (ang.).
- Varsha J. Galani, Digvijay G. Rana, Depression and antidepressants with dopamine hypothesis-A review, „International Journal of Pharmaceutical Frontier Research”, 1 (2), 2011, s. 45–60 (ang.).
- O. Benkert i inni, Altered tyrosine daytime plasma levels in endogenous depressive patients, „Archives of General Psychiatry”, 25 (4), 1971, s. 359–363, DOI: 10.1001/archpsyc.1971.01750160071013, PMID: 5116991 (ang.).
- W Birkmayer, Linauer W, Storung D. Tyrosin and tryptophan- metabolisms in depression patients. „Arch Psychiar Nervenkr”. 213, s. 377–387, 1970 (ang.).
- M.B. Bowers, G.R. Heninger, F. Gerbode, Cerebrospinal fluid 5-hydroxyindoleacetic acid and homovanillic acid in psychiatric patients, „International Journal of Neuropharmacology”, 8 (3), 1969, s. 255–262, DOI: 10.1016/0028-3908(69)90046-x (ang.).
- Jeffrey N. Carlson i inni, Chronic antidepressant drug treatment reduces turning behavior and increases dopamine levels in the medial prefrontal cortex, „Brain Research”, 707 (1), 1996, s. 122–126, DOI: 10.1016/0006-8993(95)01341-5 (ang.).
- G.G. Nomikos i inni, Acute effects of bupropion on extracellular dopamine concentrations in rat striatum and nucleus accumbens studied by in vivo microdialysis, „Neuropsychopharmacology”, 2 (4), 1989, s. 273–279, PMID: 2482026 (ang.).
- Artykuł grupy psychiatrów amerykańskich na temat potencjału przyłączania się dopaminy do receptorów D2http://ajp.psychiatryonline.org/cgi/content/full/157/3/457
- F.R. Schneier i inni, Low dopamine D(2) receptor binding potential in social phobia, „The American Journal of Psychiatry”, 157 (3), 2000, s. 457–459, DOI: 10.1176/appi.ajp.157.3.457, PMID: 10698826.
W źródła wliczają się również przypisy i bibliografia wszystkich artykułów kategorii: "Neuroprzekaźniki" Wikipedii. Przeglądałem te źródła, tłumaczyłem ich fragmenty. Artykuł jest skondensowaną formą przekazania wiedzy zawartych w tych różnorodnych źródłach. Nad artykułem pracowałem ponad dwa tygodnie. Mam nadzieję, że będzie on Wam pomocny w przyswojeniu wiedzy na temat neuroprzekaźników:

Niezależnie od tego, czy borykasz się z depresją, lękiem czy innym zaburzeniem psychicznym, pomoc psychiatryczna jest niezbędna do poprawy twojego stanu zdrowia. Odwiedź stronę https://psycholog-ms.pl/pomoc-psychiatryczna/, aby dowiedzieć się więcej o dostępnych opcjach terapeutycznych.
OdpowiedzUsuńWsparcie psychologiczne może być potrzebne w różnych sytuacjach życiowych. Strona https://www.terapiapoznan.pl/ może zawierać informacje o specjalnych programach terapeutycznych dla różnych grup klientów, jak np. terapia rodzinna czy wsparcie dla osób w żałobie.
OdpowiedzUsuń